Structural and Electronic Properties of Folic Acid Adsorption on the Carbon Nanotubes: A Density Functional Theory Study
Shahla Hamedani
Department of Chemistry, Science and Research Branch, Islamic Azad University, Tehran, Iran
DOI : http://dx.doi.org/10.13005/ojc/310140
Article Received on :
Article Accepted on :
Article Published : 02 Mar 2015
In this study, we studied the adsorption behavior of folic acid on the (5,0) zigzag and (5,5) armchair single-walled carbon nanotube (SWCNT) by Density Functional Theory (DFT). Geometry optimizations were carried out at the B3LYP/6-31 G* level of theory using the Gaussian 09 suite of programs. The adsorption energies, the quantum molecular descriptors analysis and the structural changes at the adsorption site are indicative of chemisorption (adsorption energy -31.673 kcal/mol) on the zigzag SWCNT surface, while the adsorption is physical (adsorption energy -9.70 kcal/mol) in nature on the armchair SWCNT surface. The density of the states (DOS) Plots, Natural bond orbital (NBO) analysis, and HOMO and LUMO are witness to the substantial changes in the electronic properties of the SWCNT systems after the attachment adsorbed species with the tube surface. These results are extremely relevant in order to diagnosis the potential applications of carbon nanotubes as drug delivery systems.
KEYWORDS:Structural and Electronic; studied; chemisorption
Download this article as:| Copy the following to cite this article: Hamedani S. Structural and Electronic Properties of Folic Acid Adsorption on the Carbon Nanotubes: A Density Functional Theory Study. Orient J Chem 2015;31(1). |
| Copy the following to cite this URL: Hamedani S. Structural and Electronic Properties of Folic Acid Adsorption on the Carbon Nanotubes: A Density Functional Theory Study. Orient J Chem 2015;31(1). Available from: http://www.orientjchem.org/?p=7518 |
Introduction
Single-walled carbon nanotubes (SWCNTs) are formed by the rolling of a single layer of sp2 carbon, called a graphene sheet, into a seamless hollow cylindrical tube with Nano scale dimensions.Graphene makes strong σ bonds with the other 3 identical carbon atoms at an angle of 120 leaving a weekly π bonded pz electrons[1-5].CNTs are being examined as one of the most promising transfection vectors for drug and gene delivery, due to their large surface area, stability, flexibility and bio compatibility. Recently the study of the interactions of biological molecules with SWCNTs has considered because of the importance of such compounds as carriers in drug delivery systems [6, 7].
Folate and folic acid (C19H19N7O6) are forms of a water-soluble B vitamin. Folate occurs naturally in food, and folic acid is the synthetic form of this vitamin. Folic acid deficiency is considered to be one of the most common nutritional deficiencies. It is used for memory loss, Alzheimer’s disease, preventing and treating low blood levels of folate, as well as its complications, including “tired blood” (anemia), preventing the eye disease, osteoporosis, sleep problems, depression, nerve pain, muscle pain, AIDS. It is also used for reducing harmful side effects of treatment with the medications lometrexol and methotrexate. since folic acid is an attractive ligand, it is useful for targeting cell membranes and enhancing CNTs endocytosis by the folate receptor [8, 9].
The objective of the present work was to study the interaction of folic acid drug with (5,0) zigzag and (5,5) armchair SWCNTs by performing density functional theory calculations.Also, the secondary aim of this study was to investigate the electronic properties such as quantum molecular descriptors, atomic charges, HOMO and LUMO energies,… of FA/SWCNT complex.
Computational Method
The two single-walled CNT models considered here are (5, 5) armchair and (5,0) zigzag with their ends saturated by hydrogen atoms. The (5, 5) model is consisted of 80 atoms, and the (5,0) model is formed of 50 atoms.All geometries have been fully optimized at the DFT/B3LYP/6-31 G* level of theory [10]by using the Gaussian 09 [11] program suite.
The frequency calculations were applied at the same level of the theory to confirm that the structures obtained corresponded to energy minima. Mulliken charges, adsorption energies, molecular electrostatic potential (MEP) analyses and density ofstates (DOS) analysis have been calculated on the SWCNT and FA/SWCNT complexe using DFT method. Also, GaussSum program [12] has been applied to obtain the DOS results.
The interaction energy (Ead) of the adsorbate molecule with the SWCNT is calculated as follows:
Ead =E FA/SWCNT – (ESWCNT + EFA) (1)
where, EFA/SWCNT is the optimized energy of the molecule folic acid adsorbed on the SWCNT system, ESWCNT are the optimized energies of the SWCNT[(5,5) and (5,0)] and EFA is the optimized energy of the molecule folic acidto calculate the adsorption energy was used the basis set superposition error (BSSE) have been estimated using the counterpoise (CP) method [13].By definition, a negative value of Ead corresponds to exothermic adsorption.
For all the optimized geometries, the Quantum Mechanical Descriptors (QMD) [14, 15]used to describe the electronic properties such as, ionization potentials (I)[16], chemical potential (μ), energy gap (Eg), electrophilicity index (ω) [17]{Parr, 1999 #2411}, global hardness (η), global softness (S) and electronegativity (χ),Electron affinity(A)[18],and ∆N [19]. These have been given by the expression:
[μ= −χ = − (I + A)/2], [η = (I − A)/2], [ω = μ2/2η], and [S = 1/2η], [I= – EHOMO], [A= – ELUMO](2)
The global interaction between the SWCNT [(5,5) and (5,0)], with folic acid molecule can be shown by the parameter ΔN, which determines the fractional number of electrons, transferred from a system A (folic acid) to another system B (SWCNT) and has been calculated as follows:
ΔN= (µB-µA) / 2(ηA+ηB) (3)
where, μA, μB and ηA, ηB are the chemical potential and chemical hardness of the systems A and B, respectively.when the charge flows from B to A the value of ΔN is positive and the A acts as an electron acceptor, while a negative value of ΔN shows that charge flows from A to B and A acts as an electron donor.
Results and Discussion
The optimized geometrical structures of pristine (5,5) armchair and (5,0) zigzag single-walled CNT at the B3LYP/6-31G* computational level are depicted in Fig. 1.Since the charge density of the oxygen atoms of the folic acid molecule is more focused, therefore, it can approach to the armchair and zigzag SWCNT surface with different orientations.
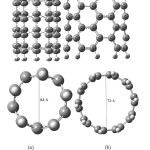 |
Figure1: Optimized geometrical structures of (a) (5,0)- zigzag SWCNT, (b ) (5,5) armchairSWCNT. Click here to View figure |
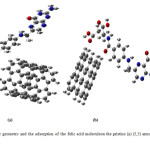 |
Figure2: Molecular geometry and the adsorption of the folic acid moleculeon the pristine (a) (5,5) armchair and (b) (5,0) zigzag SWCNT. Click here to View figure |
Prediction of real interaction energy of the adsorbate with the adsorbent consist of description of intermolecular parameters such as adsorption energies, dipole moments and equilibrium distance between the adsorbent and adsorbate are presented in Table 1.
Table 1.Calculated adsorption energies (∆E ads), equilibrium tube-molecule distance (R),total energy(Etotal),and dipole moment of the folic acidadsorbed on zigzag (5,0) and armchair (5,5), Nanotubes
|
Model |
R (Å) |
Diameter(Å) |
Etotal(kcal mol-1) |
∆E ads(kcal mol-1) |
µD |
| (5,5)-SWCNT |
– |
7.1 |
-1434310.296 |
– |
0.00 |
| (5,5)SWCNT-FA |
2.40 |
7.1 |
-2414160.017 |
-9.700 |
10.980 |
| (5,0)- SWCNT |
– |
4.1 |
-954588.3204 |
– |
0.9939 |
| (5,0) SWCNT- FA |
2.14 |
4.1 |
-1934460.015 |
-31.673 |
18.615 |
Table 2. Quantum molecular descriptors for the FA molecule, (5,0) and (5,5) SWCNT and FA drug molecules with SWCNT in gas phase. All values are in units of eV
|
Molecular descriptors |
(5,0) SWCNT |
(5,5) SWCNT |
Folic Acid |
||
|
SWCNT |
FA-SWCNT |
SWCNT |
FA-SWCNT |
||
|
E HOMO |
-3.728 |
-3.677 |
-4.387 |
-4.083 |
-5.100 |
|
ELUMO |
-2.994 |
-2.379 |
-2.738 |
-2.423 |
-2.060 |
|
ELUMO – EHOMO |
0.734 |
1.298 |
1.649 |
1.660 |
3.04 |
|
Ionization energy EI= -EHOMO |
3.728 |
3.677 |
4.387 |
4.083 |
5.100 |
|
Electron affinity EA= -ELUMO |
2.994 |
2.379 |
2.738 |
2.423 |
2.060 |
|
Chemical hardness η = (I-A) /2 |
0.367 |
0.649 |
0.824 |
0.830 |
1.52 |
|
Ch. Potential µ = -(I+A) /2 |
-3.361 |
-3.028 |
-3.562 |
-3.253 |
-3.58 |
|
Electrophilicity ω = µ2/2η |
15.390 |
6.292 |
7.70 |
6.374 |
4.215 |
|
ΔN |
– |
0.058 |
– |
0.003 |
– |
As seen in this Table, it was found that the stability of FA-SWCNT complex and molecules binding energy decrease as their diameter increase[20].When folic acid molecule is adsorbed on the surface single-walled CNTs, dipole moment of the complex significantly increased about 10.980 in (5,5) SWCNT- FA to 18.615 in (5,0) FA/SWCNT complex (Table 1). This effect can be understood by polarization of the conducting electrons due to physisorption and chemisorption.
For understand the nature of interaction between SWCNT and FA drug, we studied the electronic properties of the complex. The highest occupied molecular orbital (HOMO), lowest unoccupied molecular orbital (LUMO) and energy gaps (Eg) in FA molecule, pristine (5,5) armchair and (5,0) zigzag SWCNT and FA molecule loaded on SWCNT are depicted in Fig. 3.
The quantum molecular descriptors forFA molecule, (5,0) and (5,5) SWCNT and FA/SWCNT complex are summarized in Table 2.We observe that in the complex the energy gap increases in molecular orbitals. The increase of the energy gap by the functional group may be able to decrease the reactivity of the FA attached (5,5) and (5,0) SWCNT complex, and shows a charge transfer to take place between the (5,5) and (5,0) SWCNT surface and the FA molecule.
As observed in Table 2, increasing the energy gap causes increasing ionization potential and global hardness of the complex, that these because of the functional group proposes the increasing of stability and decrease in reactivity of the FA/SWCNT complex. The global electrophilicity index is a measure of the electrophilic power of the system. Higher is the electrophilicity index greater is its tendency to behave as an electrophile. The electrophilicity of the complex is lower than of the pristine (5,5) armchair and (5,0) zigzag SWCNT. Therefore, folic acid adsorption on the SWCNT decreases the electrophilicity of the single-walled carbon nanotube.
The calculated energy gap of the pristine (5,0) and (5,5) SWCNT are 0.734 and 1.649 eV respectively, whereas in the complexes these values increase to 1.298 and 1.660, respectively. These results indicate that FA molecule adsorption on the pristine (5,0) zigzag SWCNT has more influences on the energy gap than the pristine (5,5) armchair SWCNT. Moreover, as the global hardness of the SWCNTs are lower than that of the FA molecule attached (5,5) armchair and (5,0) zigzag SWCNT, we can predict that the complexs are relatively stable and the FA molecule adsorption process is dominant.To better understanding of the interaction between the adsorbent surface and adsorbate, the amount of charge transfer as calculated using the ΔN method, is given in Table 2{Baei, 2011 #2415}[21].
In the FA/SWCNT complexes, the ΔN values are positiveindicating that the FA molecule acts as an electron acceptor (see Table 2). Thus, electrons will flow from a definite occupied orbital in a SWCNT and will go into a definite empty orbital in a folic acid molecule.
 |
Figure3: Orbital depiction of HOMO and LUMO in (5,5), (5,0) SWCNT, and FA drug molecules optimized at B3LYP/6-31G* level of theory. Click here to View figure |
In order to examine the changes of electronic properties of SWCNT, it is essential to calculate the density of state (DOS) plots of the SWCNTs after folic acid molecule adsorption. DOS plots for the combined system of FA/SWCNTs compared with the corresponding DOS for the individual parts FA and SWCNTs separated were done, as shown in Fig. 4.
 |
Figure4: DOS and band structure of a) folic acid, b) (5,0) zigzag SWCNT, c) (5,5) armchair SWCNT,d) FA/(5,0) SWCNTcomplex, and e) FA/(5,5) SWCNT complex. Click here to View figure |
Conclusion
Density functional theory calculations were used for investigate the structural and electronic changes in the armchair and zigzag SWCNT systems for adsorption of FA molecule on the SWCNT. From the amount of energy absorbed, structural change and energy gap, it was found that the absorption FA molecule on the (5,0) SWCNT is much stronger than that with (5, 5) SWCNT. In addition, the increase in global hardness and energy gap suggest the increasing of stability and decrease reactivity of the FA/SWCNT complex. In the FA/SWCNT complex, ΔN values are positive, indicating that the FA acts as electron acceptor. Interestingly, NBO analysis showed that the interaction of nanotubes with FA is more electrostatic in nature.
References
- Saito, Y., et al., Field emission from multi-walled carbon nanotubes and its application to electron tubes. Applied Physics A: Materials Science & Processing, 1998. 67(1): p. 95-100.
- De Heer, W.A., A. Chatelain, and D. Ugarte, A carbon nanotube field-emission electron source. Science, 1995. 270(5239): p. 1179-1180.
- Collins, P.G., et al., Nanotube nanodevice. Science, 1997. 278(5335): p. 100-102.
- Nardelli, M.B., B. Yakobson, and J. Bernholc, Mechanism of strain release in carbon nanotubes. Physical review B, 1998. 57(8): p. R4277.
- Huang, J., et al., Real-time observation of tubule formation from amorphous carbon nanowires under high-bias Joule heating. Nano letters, 2006. 6(8): p. 1699-1705.
- Kang, B., et al., Cancer‐cell targeting and photoacoustic therapy using carbon nanotubes as “Bomb” agents. Small, 2009. 5(11): p. 1292-1301.
- Xia, W. and P.S. Low, Folate-targeted therapies for cancer. Journal of medicinal chemistry, 2010. 53(19): p. 6811-6824.
- Zhang, X., et al., Targeted delivery and controlled release of doxorubicin to cancer cells using modified single wall carbon nanotubes. Biomaterials, 2009. 30(30): p. 6041-6047.
- Castillo, J.J., et al., Detection of cancer cells using a peptide nanotube–folic acid modified graphene electrode. Analyst, 2013. 138(4): p. 1026-1031.
- Becke, A.D., Density‐functional thermochemistry. III. The role of exact exchange. The Journal of Chemical Physics,1993. 98(7): p. 5648-5652.
- Frisch, M., Gaussian 09 (Revisions A. 02). 2009, Gaussian, Inc., Walling ford CT, USA.
- O’boyle, N.M., A.L. Tenderholt, and K.M. Langner, Cclib: a library for package‐independent computational chemistry algorithms. Journal of computational chemistry, 2008. 29(5): p. 839-845.
- Van Duijneveldt, F.B., J.G. van Duijneveldt-van de Rijdt, and J.H. van Len the, State of the art in counterpoise theory. Chemical Reviews, 1994. 94(7): p. 1873-1885.
- Chattaraj, P.K., U. Sarkar, and D.R. Roy, Electrophilicity index. Chemical reviews, 2006. 106(6): p. 2065-2091.
- Hazarika, K.K., N.C. Baruah, and R.C. Deka, Molecular structure and reactivity of antituberculosis drug molecules isoniazid, pyrazinamide, and 2-methylheptylisonicotinate: a density functional approach. Structural Chemistry, 2009. 20(6): p. 1079-1085.
- Roy, R.K. and S. Saha, Studies of regioselectivity of large molecular systems using DFT based reactivity descriptors. Annual Reports Section” C”(Physical Chemistry), 2010. 106: p. 118-162.
- Parr, R.G., L.v. Szentpaly, and S. Liu, Electrophilicity index. Journal of the American Chemical Society, 1999. 121(9): p. 1922-1924.
- Koopmans, T., Ordering of wave functions and eigenenergies to the individual electrons of an atom. Physica, 1933. 1(1): p. 104-113.
- Parr, R.G. and R.G. Pearson, Absolute hardness: companion parameter to absolute electronegativity. Journal of the American Chemical Society, 1983. 105(26): p. 7512-7516.
- Johnson, M., et al., Neutron diffraction and numerical modelling investigation of methane adsorption on bundles of carbon nanotubes. Chemical Physics, 2003. 293(2): p. 217-230.
- Baei, M.T., et al., Adsorption properties of N 2 O on (6, 0),(7, 0), and (8, 0) zigzag single-walled boron nitride nanotubes: a computational study. Computational and Theoretical Chemistry, 2011. 970(1): p. 30-35.

This work is licensed under a Creative Commons Attribution 4.0 International License.
![]()
A New Edition of Web of Science
Journal Impact Factor
2022: 0.5
Five Year: 0.8
Journal is Indexed in

Cabells Whitelist
![]()


