The Investigation of Different Properties of Clonidine Drug Binding to Carbon Nanotube: A Theoretical Study
Z. Yousefian
Department of Chemistry, Science and Research Branch, Islamic Azad University, Tehran, Iran.
DOI : http://dx.doi.org/10.13005/ojc/300428
Article Received on :
Article Accepted on :
Article Published : 18 Dec 2014
In this study, we investigated the binding of Clonidine Drug (C9H9Cl2N3) with zigzag single walled Carbon Nanotubes (SWCNTs) (5, 0) and a length of 5ᵒA by theoretical methods of theory (NMR,NBO, HOMO- LUMO Gap energy,…calculations) using Gaussian 09 software package. Then, Simulation was done in MM+, AMBER and OPLS force fields by Monte Carlo method in HyperChem. Three important energy parameters – Potential Energy, Kinetic Energy and Total Energy-calculated in five different simulating temperatures (308, 310, 312, 314 and 316 Kelvin) were used for computation and good results were obtained.
KEYWORDS:Carbon Nanotube; Clonidine Drug; Different Properties
Download this article as:| Copy the following to cite this article: Yousefian Z. The Investigation of Different Properties of Clonidine Drug Binding to Carbon Nanotube: A Theoretical Study. Orient J Chem 2014;30(4). |
| Copy the following to cite this URL: Yousefian Z. The Investigation of Different Properties of Clonidine Drug Binding to Carbon Nanotube: A Theoretical Study. Available from: http://www.orientjchem.org/?p=6001 |
Introduction
Nano technology is an advancing method with many ways for unlocking problems, especial in medical science. By performing more research on this technology, treat can be found for diseases that have no cures until now. Therefore, nanotechnology can effect on life.
One of the exciting classes of nano materials is Carbon Nanotubes (SWCNTs), which possess characteristics suitable for many applications as delivery vehicles of biologically important molecules in view of possible biomedical applications, such as vaccination and gene or Drug delivery. A useful devise to achievement these purposes is theoretical methods [1, 2].
Ideally, the Nanotubes will locate to a specific site in the body, through its functionalized surface, and release its contents. The major advantage with this form of targeted Drug delivery is the possibility of reducing the many adverse side effects experienced by patients [3-8] hence throughout various field of science and technology, a push towards the use of Nano- scale technology such as Single wall Carbon Nanotubes is on the move. One area where SWCNTs work is already well under way is within the field of Drug delivery. SWCNTs make possible bonding to Drugs [9].
One of the goal of this study was to examine the binding of Clonidine Drug with zigzag single walled Carbon Nanotubes (SWCNTs) with (5, 0) structure and a length of 50A and investigate different parameters of Drug-SWCNT complex[10].
The secondary goal of this study was investigate of Energy parameters of Drug-SWCNT complex.
Methodology
In our model, the firstly, Clonidine Drug was attached covalently to Carbon Nanotube(SWCNTs) with (5, 0) structure and a length of 5ᵒA.
All calculations were performed using Gaussian 09 software package. Geometrical optimizations of Drug, single point calculation and NMR parameters were carried out in gas phase with the Hartee -Fock method coupled to 6-31 g* basis set for all atoms.
The most common type of ab initio calculation is called a Hartee- Fock calculation (abbreviated HF), in which the primary approximation is called the central field approximation. A method, which avoids making the HF mistakes in the first place, is called Quantum Monte Carlo (QMC). These methods work with an explicitly correlated wave function and evaluate integrals numerically using a Monte Carlo integration [11, 12]. In general, ab initio calculations give very good qualitative results and can give increasingly accurate quantitative results as the molecules in question become smaller [13]. There are three steps in carrying out any quantum mechanical calculation in HyperChem. 7.0 program package [14].
First, prepare a molecule with an appropriate starting geometry. Second, choose a calculation method and its associated options. Third, choose the type of calculation
with the relevant options. For example we calculated H, C, N, Cl NMR spectral parameters for the interaction of Clonidine Drug with SWCNT in gas phase by the HF/6-31g*method.
(σ11 ≤ σ22 ≤ σ33 (1
The values of the shielding tensor are frequently expressed as the isotropic and anisotropic parts (σiso and σaniso) and the shielding asymmetry (η). [15, 16]
Geometrical optimizations of Drug were carried out with the Hartee -Fock method coupled to 6-31g basis sets for all atoms. Also, in this study we use chem Office software (chem3D and chem draw) and hyper chem at the end data will be presented as tables and figs. Simulation was done in MM+, AMBER and OPLS force fields. Molecular Mechanics calculations were assessed by Monte Carlo method [17]. Three important energy parameters – Potential Energy, Kinetic Energy and Total Energy- in five different simulating temperatures (308, 310, 312, 314 and 316 Kelvin) were used for computation.
Results and Discussion
Molecular Geometry
Fig. 1, Shows the graphical representations of the optimized geometry of Drug–SWCNT. In the figure, the Cl atoms are shown by green colors, white spheres are H atoms, blue sphere is N and gray sphere is C. Selected geometrical parameters for Clonidine Drug SWCNT are also shown in Fig.1.a.
 |
Fig1: Optimized geometries of a)Clonidine, b) Clonidine Drug-SWCNT obtained at HF/6-31g level. |
Nuclear Magnetic Resonance Parameters
NMR is based on the quantum mechanical property of nuclei. The chemical shielding refers to the phenomenon, which is associated with the secondary magnetic field created by the induced motions of the electrons that surrounding the nuclei when in the presence of an applied magnetic field [18]. In general, the electron distribution around a nucleus in a molecule is more spherically symmetric. Therefore, the size of the electron current around the field, and hence the size of the shielding, will depend on the orientation of the molecule within the applied field B0.
In the present paper, total dipole moments of Drug interaction with SWCNT in gas phase have been explored and NMR computations were done by Gaussian 09 suite of programs. The calculated magnetic shielding tensor (σ ,ppm), shielding asymmetry (η) and the chemical shift tensor (δ) calculated for C, H, N and Cl nuclei in the active site of Clonidine Drug and for carbon atoms of the open end of a SWCNT system in gas phase are presented in Table 1. Also, the graphs of calculated isotropic magnetic shielding constants σiso (ppm), anisotropic magnetic shielding tensors σaniso (ppm), Chemical shifts δ (ppm) and shielding asymmetry (η) versus the number of atomic centers for selected atoms of Drug -SWCNT system are displayed in Figs. 2a-c respectively.
As was expected, the NMR shielding tensors of H, C, N, and Cl nuclei are drastically affected by the atom to which they are bonded and by the type of the bond to the neighboring atom. The results obtained give strong evidence that intermolecular interactions play a very important role in determining the H, C, N and Cl NMR chemical shielding tensors. Some systematic trends appeared from the analysis of the calculated values.
According to Figure2a, it is obvious that one atom in Drug-SWCNT system has maximum value in compare to the other atoms of this structure and this value belongs to 33Cl. Anisotropic chemical shielding is one of the other parameters that were checked in this work. From Figure 2b it has been found that the maximum value of σaniso in Drug-SWCNT system is related to 16C. The results of investigating chemical shift tensor indicate that 32Cl have been shown to be the largest value of (δ) in system as Drug interacted with SWCNT and our knowledge about Drug interacted to SWCNT has been specified that C number 31 show the largest intermolecular effects in (η) component (Fig. 2d).
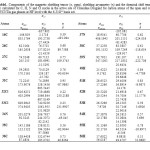 |
Table1: Components of the magnetic shielding tensor (σ, ppm), shielding asymmetry (η) and the chemical shift tensor (δ) calculated for C, H, N and Cl nuclei in the active site of Clonidine Drug and for carbon atoms of the open end of a SWCNT in gas phases at HF level with the 6-31G* basis set. Click here to View table |
 |
Fig2: The graphs of a) , b) σaniso , c) δ , d) η of propose atoms of Drug binding to SWCNT in gas phases at the HF/6-31G* basis set.
|
Natural Bond Orbital (NBO) Analysis
The concepts of natural atomic orbital NAO and NBO analyses are useful or distributing electrons into atomic and molecular orbitals used for the one-electron density matrix to define the shape of the atomic orbitals in the molecular environment and then derive molecular bonds from electron density between atoms. In NBO analysis, the input atomic orbital basis set is transformed via natural atomic orbitals (NAOs) and natural hybrid orbitals (NHOs) into natural bond orbitals (NBOs). The NBOs obtained in this fashion corresponds to the widely used Lewis picture, in which two-center bonds and lone pairs are localized [19, 28].
The NAOs will normally resemble the pure atomic orbitals and may be divided into a natural minimal basis, corresponding to the occupied atomic orbitals for the isolated atom and a remaining set of natural Rydberg orbitals based on the magnitude of the occupation numbers. The minimal set of NAOs will normally be strongly occupied, while the Rydberg NAO usually will be weakly occupied. There are as many NAOs as the size of the atomic basis set and the number of Rydberg NAOs thus increases as the basis set is enlarged.
Natural orbitals are used in computational chemistry to calculate the distribution of electron density on atoms and in bonds between atoms. NBOs include the highest possible percentage of the electron density, ideally close to 2.000 [20-22]. This is carried out by considering all possible interactions between filled donor and empty acceptor NBOs and estimating their energetic importance by second-order perturbation theory. For each donor NBO (i) and acceptor NBO (j), the stabilization energy E 2 associated with electron delocalization between donor and acceptor is estimated as

Where qi is the orbital occupancy, εi, εj are diagonal elements and Fi, j is the off-diagonal NBO Fock matrix element [21].
The aim of the present work is to investigate the nature of bonding in ClonidineDrug- SWCNT using natural bond orbital (NBO) analysis. We have shown that the results from NBO calculations can provide the detailed insight into the electronic structure of molecule. The results of NBO analysis at HF/6-31G* level of theory are listed in Table2.
These tables summarize the second-order perturbative estimates of ‘donor- acceptor’ interactions. This analysis is carried out by examining all possible interactions between ‘filled’ (donor) Lewis-type NBOs and ’empty’ (acceptor) non-Lewis NBOs, and estimating their energetic importance by 2nd-order perturbation theory. Since these interactions lead to loss of occupancy from the localized NBOs of the idealized Lewis structure into the empty non-Lewis orbitals (and thus, to departures from the idealized Lewis structure description), they are referred to as ‘delocalization’ corrections to the zeroth-order natural Lewis structure. For each donor NBO (i) and acceptor NBO (j), the stabilization energy E 2 associated with delocalization (“2e-stabilization”) is estimated.
According to Table2, it is obvious that one σ bond in Drug-SWCNT system has maximum Occupancy value in N37-C38.
Also, F (i, j), E2 and E (j)-E (i)define Correlation Energy and Coefficients Hybrids is one of the other parameters that are checked in this work.Also, strongest interaction in these compounds are identified for the interaction of BD (1) → BD* (1) in Drug-SWCNT system.
 |
Table2: NBO analysis of Drug binding to SWCNT in gas phases at HF level with the 6-31G*basis set. Click here to View table |
Electromagnetic Hyperfine Parameters
In this section, the major point is embedded in the investigation of the electrostatic interaction of Clonidine Drugwith the open end of a SWCNT in gas phase by the HF/6-31g*method. Length bonds, total atomic charges, Electric Potential in different bonds of Drug and Drug-SWCNT system are reported in Tables3, 4. Also, graphs of calculated electric potential in different bonds of Drug and Drug-SWCNTsystem in Figs. 3, 4.The calculations were performed in two methods and different electrostatic properties.
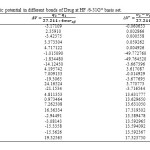 |
Table3: Electric potential in different bonds of Drug at HF /6-31G* basis set. Click here to View table |
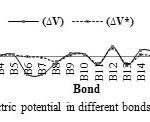 |
Fig3: The graph of calculated electric potential in different bonds of Drug at the HF/6-31G* basis set. Click here to View figure |
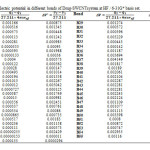 |
Table4: Electric potential in different bonds of Drug-SWCNT system at HF/6-31G* basis set. Click here to View table |
 |
Fig4: The graph of calculated electric potential in different bonds of Drug-SWCNT system at the HF/6-31G* basis set.
|
HOMO, LUMO and HOMO- LUMO Gap energy
Table 5 shows the values of HOMO, LUMO, HOMO–LUMO Gap energyfor Drug and Drug-SWCNTsystem using HF with 6-31G* basis set. Table 5 demonstrates that From HOMO–LUMO Gap energy calculation, it can be seen that HOMO- LUMO Gap energy of decrease in the order: Clonidine Drug>Clonidine Drug-SWCNT system and by decreasing of HOMO- LUMO Gap energy, would be more stable compound. So, Clonidine Drug beside SWCNT can act better as an electron donor and probably all of its biochemical and molecular functions can be accounted for by this function [21-24].
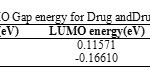 |
Table5: HOMO, LUMO and HOMO- LUMO Gap energy for Drug and Drug-SWCNT system. Click here to View table |
Calculation of Binding Energies for Clonidine Drugto SWCNT
Binding parameters such as binding energy, Enthalpy, Free Gibbs energy and Entropy are calculated for Clonidine Drug and Clonidine Drug to SWCNT. The results are shown in table 6. According to the frequency calculation at the HF/6-31G* level of theory, connection of SWCNT to Clonidine Drug is a weak connection. Also, results in Table 6 indicate that energy (ΔE) and enthalpies (ΔH) values as well as free Gibbs energies (ΔG) obtained are negative, signifying that such interaction is favorable thermodynamically.
 |
Table6: Binding energies for Drug to SWCNT at the HF/6-31G* level. Click here to View table |
Energy Parameters
In current study computations were done in sophisticated and appropriate molecular modeling environment of Hyper Chem which is well known for its quality and flexibility [25,26]. It is known that atoms are held together by forces. Function of biological systems arises from interaction of resilient bonds between atoms and electron motion. The main purpose is to seek for the lowest energy, in which the molecule is in its most stable state [27, 28]. In this study AMBER, MM+ and OPLS force fields were chosen. The total Potential Energy is the sum of mentioned contribution interactions based on the force fields.
Therefore, force fields are a series of functional energy parameters that evaluate performance and calculate the Potential Energy of molecule in various positions of its constituent atoms and bonds [29].
MM+ is a proper parameter for attaining vibration motion of atoms, related bond stretching potential, and angles bending. AMBER force field has extensive application for proteins and nucleic acids. It assigns all conformational energies and treats with hydrogen bond energy, and torsion term [30]. Like AMBER, OPLS is designed for computation of proteins and nucleic acids. In this force field bonded potentials are similar to AMBER and its non-bonded potentials involve vander Waals and electrostatics. Similar to AMBER and OPLS it has been designed to study macromolecules.
Clonidine Drug to Carbon Nanotube was simulated in mentioned force fields in 5 different temperatures (308K, 310K, 312K, 314K and 316K). To elucidate the effect of Clonidine Drug to Nanotube energy on molecular mechanic calculation, the most usual expression for total potential energy is given by the following equation:

E total is the sum of bonded and non-bonded interactions
E bond is stretching bond energy between two atoms
Eangle is energy of bending an angle
E torsion is torsion energy of rotation around a bond
E electrostatic and E van der Waals are two energies which are exponent distribution, and repulsion or attraction between non-bonded atoms, respectively.
The other two calculated energy quantities are kinetic and total energy values. In symbols the total energy equals:

From a statistical point of view, the obtained valuable data for three basis sets of thermodynamic parameters (E potential, E kinetic, E total), analyzed under the different simulation procedure, various temperatures values every 10 (PS) span are listed in tables7, 8 and 9.
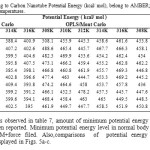 |
Table7: Computed Drug to Carbon Nanotube Potential Energy (kcal/ mol), belong to AMBER; MM+ and OPLS force fields in five different temperatures. Click here to View table |
According to results observed in table 7, amount of minimum potential energy calculated by MM+ force filed have been reported. Minimum potential energy level in normal body temperature (310K) was 316.2 for MM+force filed. Also,comparisons of potential energy levels in different temperatures are displayed in Figs. 5a-c.
 |
Fig5: The graphs of Drug-SWCNT Potential Energy a)MM+, b) OPLS, c) AMBER in Monte Carlo method. |
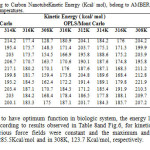 |
Table8: Computed Drug to Carbon Nanotube Kinetic Energy (Kcal/ mol), belong to AMBER, MM+ and OPLS force fields in five different temperatures. Click here to View table |
It is known that to have optimum function in biologic system, the energy levels must be in the minimum level. According to results observed in Table 8and Fig.6, for kinetic energy in different time steps and various force fields were constant and the maximum and minimum quantity observed in 310K, 285.5Kcal/mol and in 308K, 123.7 Kcal/mol, respectively.
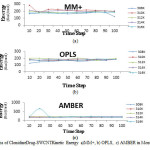 |
Fig6: The graphs of Clonidine Drug-SWCNT Kinetic Energy a)MM+, b) OPLS, c) AMBER in Monte Carlo method. Click here to View figure |
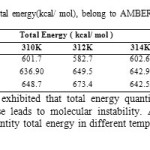 |
Table9: Computed Drug-SWCNT Total energy(kcal/ mol), belong to AMBER, MM+ and OPLS force fields in five different temperatures. Click here to View table |
Also, data analysis of table 4 exhibited that total energy quantities were affected by increasing temperature that energy increase leads to molecular instability. According to results observed in Table 9and Fig.7 maximum quantity total energy in different temperature was 312K, 673.4Kcal/mol in amber method.
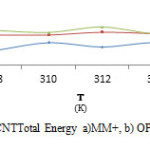 |
Fig7: The graph of Clonidine Drug-SWCNTTotal Energy a)MM+, b) OPLS, c) AMBER in Monte Carlo method. Click here to View figure |
Conclusion
- Nuclear magnetic resonance (NMR) chemical shielding tensors in the methodsframework makes it possible to study the chemical shift of Carbon Nanotubes. Chemical shift anisotropy asymmetry (η), isotropy (σ iso), anisotropy (σ aniso) and chemical shift tensor (δ) are observed for the atoms ofClonidineDrug-SWCNT. Our obtained results yielded strong evidence that intermolecular interaction effects such as electron transfer interactions play very important role in determining NMR chemical shielding tensors of the atoms are characterized in Drug-SWCNT and some systematic trends appeared from the analysis of the calculated values. The calculated parameters reveal that Cl57 and H83 atoms have the largest and smallest σaniso constants among the other nuclei, respectively. The Cl57 has the largest but N62has the smallest chemical shift (δ) constants among the other atoms, respectively. Also, the diagrams consist of σiso,σaniso and η showall shielding values but δ show more negative shielding values at the HFin 6-31G* basis set.
- In Natural Bond Orbital (NBO) analysis, orbital occupancy, F (i, j), E2 and E (j)-E (i)define Correlation Energy and Coefficients Hybrids is one of the other parameters that are checked in this work. According to Table2 it is obvious that one σ bond in Drug-SWCNT system has maximum Occupancy value in N37-C38. Also, strongest interaction in these compounds are identified for the interaction of BD (1) → BD* (1) in Drug-SWCNT system.On the basis of the constant values of the coefficients of a linear combination of s and p orbitals of different bonds were between0.6 and 0.7, a specific voltage difference could be expected.It is observed that the percent of s and p orbitals for different bonds in Drug-SWCNT system at all coordination refers to sp2 hybridization for some atoms, which is in agreement with the intrinsic sp2 hybridization of atoms.
- In addition, electric potential in different bonds of some atoms – Drug and Drug-SWCNT system are investigated. The calculations are performed in two different methods and these are shown in Table 3, 4 and Figs. 3, 4.
- We analyze the electronic structure and charge Mullikan population for the energetically most favorable complexes. Binding parameters and binding energies, HOMO, LUMO, Gap energy, ΔE, ΔH and enthalpies ΔG are calculated. The obtained large negative values of the ΔG confirmed the structural stability of Drug-SWCNT system in gas phase (results indicate that ΔE and ΔH values as well as ΔG obtained are negative, signifying that such interaction is favorable thermodynamically). From HOMO–LUMO Gap calculation, it can be seen that HOMO- LUMO Gap energy of decrease in the order: Clonidine Drug> Clonidine Drug-SWCNT system and by decreasing of HOMO- LUMO Gap energy, would be more stable compound. So, Clonidine Drug beside SWCNT can act better as an electron donor and probably all of its biochemical and molecular functions can be accounted for by this function and by decreasing of HOMO- LUMO Gap energy, would be more stable compound.
- After conducting the Molecular Mechanic study and gaining the potential energy by Monte Carlo method and studying the Nanotube that were involved with the Drug in different temperature, the following results were concluded: The study showed that the system has the different level of energy and the different stability which is caused by the forces from inside the Drug to Carbon Nanotube because this Carbon Nanotube should find the best spatial conformity which means the highest stability level or the lowest level of energy. Also, you see in above diagrams, we have maximum amount of potential energy in 308K, OPLS method and the highest level of total energy observed in amber method. So with considering high amount of total energy, there will be minimum stability in this method.
References
- Monajjemi, M.; Honaparvar, B.; Khalili Hadad, B.; Ilkhani, A. R.; Mollaamin, F. Thermo-chemical investigation and NBO analysis of some anxileotic as nano –Drugs.Afr.J.2010, 4, 8.
- Monajjemi, M.; Mousavi, M.; Rezaei, S.; Mollaamin, F. Molecular dynamic and Monte Carlo study on nano energetic binding sites of neuraminidase in different media.Afr.J.2012, 6, 8.
- Zheng, Q.; Jiang, Q. Multiwalled Carbon Nanotubes as gigahertz oscillators.Phys. Rev. Lett.2002, 88.
- Baowan, D.; Angew, Z. Math. Phys.2007, 58.
- Hillebrenner, H.; Buyukserin, F.; Stewart, J.D. Template synthesized Nanotubes for biomedical applications.Nanomedicine. 2006, 1.
- Monajjemi, M.; Mahdavian, L.; Mollaamin, F.; Khaleghian, M. Interaction of Na, Mg, Al, Si with Carbon Nanotube (SWCNT): NMR and IR Study.Russian Journal of Inorganic Chemistry.2009, 54, 9.
- Monajjemi, M.; Mahdavian, L.; Mollaamin, F. Characterization of Nano crystallinesilicon germanium film and Nanotube in adsoption gas by Monte Carlo and langevin dynamic simulation Bull. Chem. Soc.2008, 22, 2.
- Aghaie, H.; Gholami, M.R.; Monajjemi, M.; Ganji, M.D. Electron transport phenomenon simulation through the carborane nano-molecular wire Physica E: Low-Dimensional Systems and Nanostructures. 2008, 40,9.
- Ramanathan, T.; Fisher, F. T.; Ruoff, R. S.; Brinson, L. C. chem mater.2005,17, 1290.
- Monajjemi, M.; Lee, V. S.; Khaleghian, M.; Honarparvar, B.; Mollaamin, F.J. Phys. Chem. C.2010, 114.
- Liu, J.C.; Monson, P.A. Adsorption. 2005, 11, 5.
- Lee, C.; Yang, W.; Parr G.R. Phys. Rev. B. 1988,37,785.
- Young, D. Cytoclonal Pharmaceutics Inc, Introduction to Computational Chemistry.
- Hyper Chem. 7.0, Hypecube Inc., Gainesville, FL, USA, 2001.
- Monajjemi, M.; Noei, M.; Mollaamin, F. Nucleosides Nucleotides Nucleic Acids.2010, 29, 676.
- Monajjemi, M.; Rajaeian, E.; Mollaamin, F.; Naderi, F.; Saki, S. Phys. Chem. Liq.2008, 46, 3299.
- http://www.cup.uni-muenchen.
- Ghiasi, R.; Mokaram, E. Journal of Applied Chemical Research.2012, 20,7-13.
- Norberg, J.; Nilsson, L. Ac Chem Res.2002,35,6, 465-72
- http://www.huntresearchgroup.org.
- Zakharian, T.J. Am Chem Soc.2005,127, 12508-12509.
- Liu, Z. Carbon Nanotubes in Biology and Medicine: In vitro and in vivo Detection, Imaging and Drug Delivery. Nano Res. 2009, 2, 85-120.
- Liu, Z.J. Mater Chem.2011, 21, 586-598.
- Yang, W. Carbon nano materials in biosensors.Angew Chem Int Ed.2010, 49, 2114-2138.
- Lewars, E. J. Computational chemistry, Ontario Canada.2003.
- Nam, K.;Gao, J.;York, D. M.J. Am. Chem. Soc.2008,130.
- Mackerell, A. D. J. Comput. Chem.2004, 25.
- Weiner, S.J.;Kollman, P.A., et al.Journal of the American Chemical Society.1984, 106, 3.
- Foloppe, N.;Mackerell. A.J. Comput. Chem.2000, 21.
- Case,D.A., Cheatham, T., Darden, T.,Gohlke, H., Luo, R. K., Merz, M.,Onufriev, A., Simmerling, C., Wang, B., Woods. R.J. Comput. Chem.2005, 26.

This work is licensed under a Creative Commons Attribution 4.0 International License.
![]()
A New Edition of Web of Science
Journal Impact Factor
2022: 0.5
Five Year: 0.8
Journal is Indexed in

Cabells Whitelist
![]()


