NMR and NBO investigation of Dopamine properties in point view of Brain activities
M. Mehdizadeh Barforushi*
Department of Chemistry, College of Basic Sciences, Tehran science and Research Branch, Islamic Azad University, Tehran, Iran
DOI : http://dx.doi.org/10.13005/ojc/300443
Article Received on :
Article Accepted on :
Article Published : 01 Jan 2015
Dopamine dosage is the main reason for panic, fear as well as exhilaration. In this study, it has been shown that the electron negativity of Cl, Br and F ions plays an important role in binding of these ions to dopamine receptors. The nervous quartiles effective drugs have been calculated via NMR and NBO approach. Moreover, the HOMO-LUMO energy gap of dopamine and its halogenated derivatives have been calculated at the B3LYP/6-31G* level. These calculations are so important in order to investigate many other diseases since it can exhibit the ability and relation between electron acceptor represents and electron donation in any brain activities. Finally, the accuracy of calculated result has confirmed by comparing the results to the experimental data.
KEYWORDS:Dopamine; NMR; NBO;Brain activity
Download this article as:| Copy the following to cite this article: Barforushi* M. M. NMR and NBO investigation of Dopamine properties in point view of Brain activities. Orient J Chem 2014;30(4). |
| Copy the following to cite this URL: Barforushi* M. M. NMR and NBO investigation of Dopamine properties in point view of Brain activities. Available from: http://www.orientjchem.org/?p=6586 |
Introduction
Dopamine is a nervous transporter which has a key role in the central nervous system. This nervous transporter has an important role in the normal functions of brain such as learning, memorizing (storage ability in mind), controlling the noise level and adjusting character. Also do- pamine is the brain’s Cortex and this Cortex is responsible for human behaviors [1-9].
Through the decades, dopamine has been always attracted by scientists due to important roles in many diseases such as schizophrenia, Parkinson’s and depression (e.g. increase or decrease dopamine dosage in the body can cause disorder dopamine depression [10].However the structure and reactivity of this molecule has not been investigated thoroughly [11-13].
Understanding the structure, reactivity and stability of dopamine derivateis important for understanding the acting mechanism in the body since they determine the bonding ability to receptors. In this paper, the effect of electronegativity of atoms in the binding ability to receptors has been investigated in order to improve drug treatment of this common drug.For this purpose, the energy levels of dopamine molecules and electronegativity atoms such as chlorine, fluorine, bromine ions are calculated via NMR and NBO method [14-17].
Computational Methods
The structures of dopamine and halogenated derivatives were designed primarily using Chem. Bio Draw 12.0 (Scheme 1). The geometry of the systems has been optimized at the B3LYP /6-31G* computational level [18-20]. The optimization, NBO and NMR calculations of all systems are done by density functional theory (DFT) using B3 LYP method and the standard 6-31 G* basis set, by Gaussian 09 suit of programs (Gaussian 09, Revision A.02) in the gas phase [21-24].
Mullikan charges on the atoms, dipole moments, and geometry parameters such as bond lengths were also determined by the same method. The energy barrier for rotation around the tow bond in the dopamine moleculeis also assessed and the results are represented in Table1 and Figure1 [25-27].
Table 1.The calculated energy barrier of rotation around bonds C1-O10 and C2-O11
|
Rout 1 (C6-C1-O10-H21) |
Rout 2 (C3-C2-O11-H22) |
||
|
Angstroms and Degrees |
E(k(cal/mol) |
Angstroms and Degrees |
E(k(cal/mol) |
|
-15.2 |
-2.32175 |
-166.77 |
-8.09475 |
|
-0.202 |
-1.8825 |
13.22 |
-2.259 |
|
14.8 |
-2.32175 |
28.22 |
-1.69425 |
|
29.8 |
-1.69425 |
43.22 |
-0.94125 |
|
44.8 |
-0.94125 |
58.22 |
-0.251 |
|
59.8 |
-0.251 |
73.22 |
0 |
|
74.8 |
0 |
88.22 |
-0.31375 |
|
89.8 |
-0.43925 |
103.22 |
-1.19225 |
|
104.8 |
-1.44325 |
118.22 |
-1.8825 |
|
119.8 |
-3.07475 |
133.22 |
-4.518 |
|
134.8 |
-4.95725 |
148.22 |
-6.4005 |
|
149.8 |
-6.83975 |
163.22 |
-7.781 |
|
164.8 |
-8.22025 |
178.22 |
-8.4085 |
Results and Discussion
Energies
The EHOMO, ELUMO and HOMO-LUMO energygap (Eg; Δ) of dopamine and halogenated derivatives were calculated using the B3LYP method and 6-31G* basis set and the results are presented in Table 4-9 and Table S3-S8 (Supplementary materials) [28].
The energy levels are so important since the energy level for LUMO as an electron acceptor represents the ability to obtain an electron and the energy level of HOMOrepresentsthe ability to donate an electron. As shown in Table 4-9, the molecule has the lowest energy gap is -175.7456 Kcal/mol and the dopamine molecule has the largest energy gap is-130.9417 Kcal/mol [29-31].
Geometries
The structures of dopamine and halogenated derivatives were designed primarily using of Chem. Bio Draw 12.0 in scheme 1 and the obtained results were compared to other derivate at the end. The optimization, NBO and NMR calculations of the whole systems are done by density functional theory (DFT) using B3LYP method and the standard 6-31G basis set using Gaussian 09. The optimized geometrical parameters, such as Dipole moment (Debye), energy of structure formation (HF;kcal/mol) and enthalpies (ΔH), Gibbs free energy (ΔG) are listed in Table 4-9.As shown in Table 4-9,the molecule has the lowest ΔG is 72.059013Kcal/mol and the dopamine molecule has the largest ΔG is 91.416315 Kcal/mol. The molecule has the lowestΔH is 102.29162 Kcal/mol and the dopamine molecule has the largest ΔH is 122.32726 Kcal/mol.
NMR
Nuclear magnetic resonance (NMR) is very powerful tool in studying microscopic phenomena in physics, chemistry, biology and medicine. Determination of spectral parameters using the quantum-chemistry methods helps in the assignment and interpretation of the experimental data[32]. In this study, NMR has been used for determination of microscopic properties of dopamine and it halogenated derivate [33-35].
To discuss the magnitude of the shielding tensor, it is necessary to report the three principal eigenvalues of the chemical shielding anisotropy tensor (σ11, σ22, σ33)using following equations:
a) the isotropic value of the shielding tensor which is defined as:
![]()
b) the anisotropy (Δσ) of the tensor, given by:
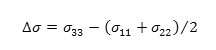
c) the shielding tensor asymmetry parameter (η) given by
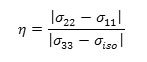
Table 2. Computed chemical shifts for selected atoms
|
structure |
atom number |
σ11 |
σ22 |
σ33 |
σiso |
Δσ |
|
C1 |
-6.7203 |
35.6456 |
129.586 |
52.8371 |
115.1234 |
|
|
-17.4526 |
17.9589 |
78.9894 |
26.4985 |
78.7362 |
||
|
-16.694 |
22.9613 |
76.7965 |
27.68793333 |
73.66285 |
||
|
-22.6423 |
57.3594 |
114.0318 |
49.58297 |
96.67325 |
||
|
3.7164 |
62.4586 |
123.6434 |
63.2728 |
90.5559 |
||
|
-25.6558 |
56.5618 |
110.3614 |
47.08913333 |
94.9084 |
||
|
11.7828 |
69.3353 |
126.8483 |
69.32213333 |
86.28925 |
||
|
C2 |
-4.2227 |
35.8531 |
130.5894 |
54.07326667 |
114.7742 |
|
|
-12.7193 |
18.0642 |
79.269 |
28.2 |
76.59 |
||
|
-8.3722 |
24.9618 |
21.9099 |
12.83316667 |
13.6151 |
||
|
-17.9397 |
59.6516 |
114.7442 |
52.15203 |
93.88825 |
||
|
8.0263 |
26.0354 |
122.4972 |
52.1863 |
105.4664 |
||
|
-20.9239 |
59.6319 |
111.1917 |
49.96656667 |
91.8377 |
||
|
29.0368 |
34.451 |
131.1433 |
64.87703 |
99.3994 |
||
|
C8 |
19.9281 |
32.2393 |
170.3295 |
74.16563333 |
144.2458 |
|
|
129.9135 |
36.9611 |
64.7332 |
143.86 |
31.29 |
||
|
130.0042 |
36.7783 |
64.3277 |
77.03673333 |
-19.06355 |
||
|
129.945 |
36.9932 |
164.9629 |
110.6337 |
81.4938 |
||
|
9.7885 |
36.8712 |
164.4894 |
70.38303 |
141.1596 |
||
|
129.7864 |
36.9806 |
165.0564 |
110.6078 |
81.6729 |
||
|
20.2364 |
31.8799 |
170.9779 |
74.36473333 |
144.91975 |
The isotropic chemical shielding σiso parameters are average of parameters, σ11, σ22 and σ33.
*Dop=dopamine
The optimization and NMR calculation are done by density functional theory (DFT) using B3LYP method and the standard 6-31G* basis set in the gas phase. Table 2 and 3 presents the computed chemical shift for selected hydrogen, nitrogen and carbon atoms. According to this Table, anisotropic shielding value (Δσ ) for C8 and N atom of F(OH)-Dopmolecule and N are negative values.Interestingly, among all of mentioned carbon atoms, C2 of F(OH)-Dophas the lowest isotropic shielding value. The κ, η and charges values are summarized in Table S1 and S2 [36-38].
Table S1. Computed chemical shifts for selected atoms
|
structure |
atom number |
η |
κ |
charge |
|
C1 |
0.552007 |
0.378372093 |
0.327738 |
|
|
0.674621696 |
0.48806174 |
0.581972 |
||
|
0.807502696 |
0.151672095 |
0.310302 |
||
|
1.241321 |
-0.17069 |
-0.07644 |
||
|
-1.6284 |
0.02036739 |
-0.111033 |
||
|
1.299425551 |
-0.208929459 |
0.048784 |
||
|
1.000457763 |
-0.000343283 |
0.047116 |
||
|
C2 |
0.52375621 |
0.405457 |
0.315797 |
|
|
0.60278251 |
0.595417964 |
0.003529 |
||
|
3.6724666 |
-1.201564621 |
0.305575 |
||
|
1.239633 |
-0.16957 |
-0.07595 |
||
|
-52.3018 |
0.685351 |
0.362558 |
||
|
1.315730904 |
-0.21947446 |
0.048189 |
||
|
0.081704 |
0.89395 |
0.328631 |
||
|
C8 |
0.128023138 |
0.836289 |
-0.128391 |
|
|
-13.8256 |
-4.052946663 |
-0.340644 |
||
|
7.335404476 |
-1.838942392 |
-0.133027 |
||
|
-1.710899479 |
6.308816348 |
-0.134595 |
||
|
-67.0237 |
0.64987 |
-0.13307 |
||
|
-1.70447 |
6.262591 |
-0.13434 |
||
|
0.1205167 |
0.845516994 |
-0.129605 |
Table S2. Computed chemical shifts for selected atoms
|
structure |
atom number |
η |
κ |
charge |
|
N |
-0.554212496 |
1.906395171 |
-0.714647 |
|
|
0.936090259 |
-3.280343256 |
-0.407913 |
||
|
3.760213469 |
-1.224908125 |
-0.715729 |
||
|
-1.86819 |
7.602459 |
-0.7154 |
||
|
-128.3092667 |
0.79741787 |
-0.715707 |
||
|
-1.86735 |
7.594614 |
-0.71544 |
||
|
-0.55607 |
1.91013 |
-0.71502 |
||
|
H19 |
0.43501 |
0.493439335 |
0.296342 |
|
|
0.634552 |
0.543629765 |
0.310717 |
||
|
0.642747705 |
0.294216612 |
0.298807 |
||
|
0.635069181 |
0.301175136 |
0.300622 |
||
|
10.1258 |
-0.417693114 |
0.298418 |
||
|
0.633387 |
0.302703 |
0.300477 |
||
|
0.432076 |
0.496426 |
0.297998 |
||
|
H20 |
0.743319474 |
0.20571089 |
0.290714 |
|
|
0.863861 |
0.199592525 |
0.2761 |
||
|
0.875599438 |
0.096295216 |
0.293655 |
||
|
0.881969434 |
0.091214448 |
0.294312 |
||
|
12.5714 |
-0.55916 |
0.293424 |
||
|
0.899 |
0.077713 |
0.294229 |
||
|
0.758255 |
0.192971 |
0.293085 |
Table 3. Computed chemical shifts for selected atoms
|
structure |
atom number |
σ11 |
σ22 |
σ33 |
σiso |
Δσ |
|
N |
95.9945 |
29.4999 |
242.7177 |
122.7373667 |
179.9705 |
|
|
191.716 |
25.3634 |
41.97 |
219.68 |
33.43 |
||
|
190.9571 |
25.0724 |
41.8411 |
85.95686667 |
-66.17365 |
||
|
191.5971 |
25.4218 |
241.9345 |
152.9845 |
133.4251 |
||
|
0.6262 |
25.0737 |
241.9851 |
89.22833333 |
229.13515 |
||
|
191.5634 |
25.4276 |
241.9487 |
152.9799 |
133.4532 |
||
|
96.2871 |
29.4256 |
243.2144 |
122.9757 |
180.3581 |
||
|
H19 |
24.7994 |
28.992 |
41.3526 |
31.71467 |
14.4569 |
|
|
23.7725 |
29.4723 |
40.0824 |
31.1 |
13.46 |
||
|
23.8236 |
29.5709 |
40.1099 |
31.16813333 |
13.41265 |
||
|
23.7761 |
29.5058 |
40.1742 |
31.15203333 |
13.53325 |
||
|
3.8093 |
29.5853 |
40.1726 |
24.5224 |
23.4753 |
||
|
23.7379 |
29.4884 |
40.2316 |
31.15263 |
13.61845 |
||
|
24.719 |
28.9215 |
41.4097 |
31.6834 |
14.58945 |
||
|
H20 |
25.6399 |
31.2533 |
39.7743 |
32.2225 |
11.3277 |
|
|
25.2384 |
31.3865 |
38.977 |
31.86 |
10.66 |
||
|
25.2569 |
31.4527 |
38.9689 |
31.89283333 |
10.6141 |
||
|
25.2443 |
31.4942 |
38.9987 |
31.9124 |
10.62945 |
||
|
5.2442 |
31.5347 |
38.9681 |
25.249 |
20.57865 |
||
|
25.2012 |
31.5541 |
38.9776 |
31.91097 |
10.59995 |
||
|
25.4854 |
31.2492 |
39.7694 |
32.168 |
11.4021 |
The isotropic chemical shielding σiso parameters are average of parameters, σ11, σ22 and σ33.
*Dop=dopamine
Table 4 . Occupancy of natural orbitals (NBOs) and hybrids of dopamine calculated by the B3LYP method with 6-31G*(d) basis set.
|
Symbol |
charge of nitrogen |
∆G |
∆H |
Energy of band gap (kcal/mol) |
E (kcal/mol) |
Dipole Moment(D |
NBO a |
Occupancy |
E |
Length(Å) |
|
-0.714676 |
91.416315 |
122.32726 |
-130.9417 |
324198.48 |
1.3422 |
1.99428 | -0.89263 | 1.367773 | ||
| 1.99428 | -0.89158 | 1.368191 | ||||||||
| 1.99352 | -0.69245 | 1.466979 | ||||||||
| 1.98953 | -0.59715 | 1.018841 | ||||||||
| 1.99081 | -0.59666 | 1.019841 | ||||||||
| 1.98816 | -0.72203 | 0.969679 | ||||||||
| 1.98812 | -0.72117 | 0.969655 | ||||||||
| CR(1) N9 | 1.99967 | -14.15973 |
– |
|||||||
| CR(1) O10 | 1.99978 | -18.97620 |
– |
|||||||
| CR(1) O11 | 1.99978 | -18.97565 |
– |
|||||||
| LP(1) N9 | 1.96300 | -0.29819 |
– |
|||||||
| LP(1) O10 | 1.98058 | -0.58666 |
– |
|||||||
| LP(2) O10 | 1.87668 | -0.30619 |
– |
|||||||
| LP(1) O11 | 1.98052 | -0.58625 |
– |
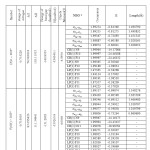 |
Table5: Occupancy of natural orbitals (NBOs) and hybrids of fluorine derivativescalculated by the B3LYP method with 6-31G*(d) basis set Click here to View table |
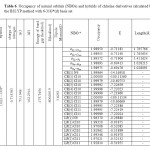 |
Table6: Occupancy of natural orbitals (NBOs) and hybrids of chlorine derivatives calculated by the B3LYP method with 6-31G*(d) basis set Click here to View table |
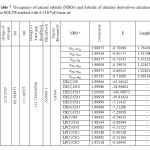 |
Table7: Occupancy of natural orbitals (NBOs) and hybrids of chlorine derivatives calculated by the B3LYP method with 6-31G*(d) basis set Click here to View table |
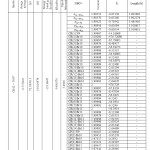 |
Table8: Occupancy of natural orbitals (NBOs) and hybrids of bromine derivatives Click here to View table |
 |
Table9: Occupancy of natural orbitals (NBOs) and hybrids of bromine derivatives Click here to View table |
NBO analysis
NBO method gives useful information about interactions in both filled and virtual orbital spaces which could enhance the analysis of intra and intermolecular interactions. This is carried out by considering all possible interactions between filled donor and empty acceptor NBOs and estimating their energy importance by second order perturbation theory [39].
NBO analysis is based on a method for optimally transforming a given wave function into localized form, corresponding to the one-center (“lone pairs”) and two-center (“bonds”) elements of the chemist’s Lewis structure picture. In NBO analysis, the input atomic orbital basis set is transformed via natural atomic orbitals (NAOs) and natural hybrid orbitals (NHOs) into natural bond orbitals (NBOs) [40-43].
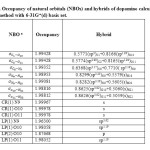 |
TableS3: Occupancy of natural orbitals (NBOs) and hybrids of dopamine calculated by the B3LYP method with 6-31G*(d) basis set. Click here to View table |
 |
TableS4: Occupancy of natural orbitals (NBOs) and hybrids of fluorine derivativescalculated by the B3LYP method with 6-31G*(d) basis set Click here to View table |
 |
TableS5: Occupancy of natural orbitals (NBOs) and hybrids of chlorine derivatives calculated by the B3LYP method with 6-31G*(d) basis set Click here to View table |
 |
TableS6: Occupancy of natural orbitals (NBOs) and hybrids of chlorine derivatives calculated by the B3LYP method with 6-31G*(d) basis set Click here to View table |
Table 4-9 lists the calculated occupancies of natural orbitals. The calculated Natural bond hybrids and are also given in this table [44-46].
Table S3-S8 show share of orbitals contribute in the bonds (BD for 2-center bond).
 |
TableS7: Occupancy of natural orbitals (NBOs) and hybrids of bromine derivatives Click here to View table |
According to calculations, the nitrogen atom forms three single bond (sigma bond) with two hydrogen atoms H19 and H20 ( σN19 -H19 and σN19 – H20), and a carbon atom C8 (σC8 – N19 ) and a lon pair orbital (Lp). As seen from Table S3-S8, the LP on the nitrogen atom is formed from an sp3.62 hybrid for the molecule, sp4.15hybrid for the molecule, sp3.63 For the molecule, sp4.15hybrid for the molecule, sp4.15hybrid For the molecule,sp3.65hybrid for the molecule, andsp3.63 hybrid For the molecule [47].
 |
TableS8: Occupancy of natural orbitals (NBOs) and hybrids of bromine derivatives Click here to View table |
Conclusion
In the present work, atheoretical analysis of dopamine and its halogenated derivate has been performed in order to obtain the dipole moment (µ) and energy of structure formation (HF)of dopamine and it halogenated derivate. Also the thermodynamic parameters of this compound has been studied and according to the results, it has been found that the amount of Gibbs free energy(ΔG) and standard enthalpies (ΔH) of dopamine and its halogenated derivatearepositive value, therefore dopamine and its halogenated derivateareunstable structure. Moreover, the chemical shifts of these molecules have been simulated using quantum mechanics.Finally, the Natural Bond Orbital (NBO) analysis has provided the detailed insight into the type of hybridization and the nature of bonding in dopamine and its halogenated derivate.
References
- Dahlstrom and Fuxe., 1964 A. Dahlstrom, K. Fuxe Localization of monoamines in the lower brain stem Experientia (1964)15, 398–399.
- Tierney et al., 2000 L. Tierney, S. McPhee, M. Papadakis, DiagnósticoClínico y Tratamiento CAP.24, 25 y 26. Quinta Edic. Edit. El Manual Moderno, 2000, pp. 965-967
- Abdel-Baki et al., 2012 A. Abdel-Baki, C. Ouetllet-Plamondon, A. Malla. Journal of Affective Disorders 138 (2012)S3.
- Santos-García et al.,2012 D. Santos-García, M. Prieto-Formoso, R. de la Fuente-Fernández, Journal of Neurological Science 318 (2012) 91.
- Monajjemi,M. Honarparvar, B. H. Haeri, H. Heshmat ,M. (2006) ,Chemistry C.,80(1):S40-S44.
- F. Mollaamin , M. Monajjemi & J. Mehrzad , Fullerenes, Nanotubes, and Carbon Nanostructures, 22: 738–751, 2014
- M. Monajjemi , N. Karachi & F. Mollaamin, Fullerenes, Nanotubes, and Carbon Nanostructures, 22(7): 643–662, 2014
- A. Tahan and M. Monajjemi,ActaBiotheor (2011) 59:291–312
- M. Monajjemi , A. Sobhanmanesh& F. Mollaamin , Fullerenes, Nanotubes, and Carbon Nanostructures, 21(1): 47–63, 2013
- Nicola et al., 2000 S.M.Nicola, J.Surmeier, R.C.Malenka, Annu Rev Neurosci, 23:185-215.
- Majid Monajjemi, Robert Wayne, JrandJames E. Boggs, Chemical Physics ,433 (2014),1-11.
- T. Ardalan , P. Ardalan& M. Monajjemi,Fullerenes, Nanotubes, and Carbon Nanostructures, 22: 687-708, 2014
- Majid Monajjemi and Fatemeh Mollaamin ,J Clust Sci (2012) 23:259–272
- M. Monajjemi ; H. Chegini ; F. Mollaamin, P. Farahani, Fullerenes, Nanotubes, and Carbon Nanostructures, 19 (5): 469–482, 2011
- F. Mollaamin , M. Monajjemi & J. Mehrzad , Fullerenes, Nanotubes, and Carbon Nanostructures, 22 (8): 738–751, 2014
- M. Monajjemi , H. Yamola& F. Mollaamin, Fullerenes, Nanotubes, and Carbon Nanostructures, 22(6): 595–603, 2014
- H. Yahyaei& M. Monajjemi,Fullerenes, Nanotubes, and Carbon Nanostructures, 22(4): 346–361, 2014
- Van Valkenburg, Jacques van der Krogt, P. Moleman, H. van Berkum, U. Tjaden,J. de Jong, Journal of Neuroscience Methods 11 (1984) 29.
- Thierry et al., 1994 A.M. Thierry, T.M.Jay, S.Pirot, J.Mantz, R.Godbout, J.Glowinski, Influence of afferent systems on the activity of the rat prefrontal cortex: Electrophysiological and pharmacological characterization. In: Motor and Cognitive Functions of the Prefrontal Cortex. (Thierry A.M., Glowinski J., Goldman- Rakic P.S., Christen Y., eds), pp 35-50. NewYork: Springer-Verlag.
- Frisch et al., 1984 M.J,Frisch, J.A.Pople, J.S.Binkley. J. Chem. Phys. 1984, 80, 3265.
- Gaussian 09, Revision A.02, M. J. Frisch et al. Gaussian, Inc., Walling ford CT, 2009.
- M. Monajjemi, J.E Boggs, J. Phys. Chem A. 117 (2013) 1670.
- Fatemeh Mollaamin and Majid Monajjemi ,J. Comput. Theor.Nanosci. 9, 597-601 (2012)
- M. Monajjemi, V. S. Lee, M. Khaleghian, B. Honarparvar, F.Mollaamin, J. Phys. Chem C., 114(2010) 15315.
- Majid Monajjemi, Chemical Physics,425 (2013),29-45.
- H. Yahyaei , M. Monajjemi , H. Aghaie, and K. Zare , Journal of Computational and Theoretical Nanoscience Vol. 10, No.10, 2332–2341, 2013.
- M. Monajjemi, Struct Chem., 23 (2012) 551-580.
- M. Monajjemi, M. Jafari Azan & F. Mollaamin,Fullerenes, Nanotubes, and Carbon Nanostructures, 21 (6) : 503–515, 2013
- M. Monajjemi,S.Afsharnezhad,M.R.Jaafari,T.Abdolahi,A.Nikosade and H.Monajemi.Russian Journal of physical chemistry A, 2007(2),1956-1963.
- Fatemeh Mollaamin & Majid Monajjemi, Physics and Chemistry of Liquids, 50 (5) 2012, 596–604
- M. Monajjemi , R. Faham & F. Mollaamin , Fullerenes, Nanotubes, and Carbon Nanostructures, 20 (2): 163–169, 2012
- Vaara 2001 J.Vaara, Theory and calculations of NMR and EPR parameters CSC Report on Scientific Coputing 1999-2000.
- M. Monajjemi ; L. Mahdavian ; F. Mollaamin ; B. Honarparvar,Fullerenes, Nanotubes and Carbon Nanostructures, 18: 45–55, 2010
- F. Mollaamin, K. Shahani pour, K. Shahani pour, A. R. Ilkhani, Z. Sheckari,d and M.Monajjemi,61(12) 2012,2193-2198 .
- M. Monajjemi , M. SeyedHosseini& F. Molaamin,Fullerenes, Nanotubes, and Carbon Nanostructures, 21: 381–393, 2013
- M. Monajjemi & M. Falahati & F. Mollaamin, Ionics (2013) 19:155–164
- F. Mollaamin, J. Najafpour, S. Ghadami, A. R. Ilkhani, M. S. Akrami, and M. Monajjemi,J. Comput. Theor.Nanosci. 11, 1290-1298 (2014)
- M. Monajjemi , N. Farahani , and F. Mollaamin , Physics and Chemistry of Liquids, 50 (2), 2012, 161–172
- Reed et al., 1988 A.E.Reed, L.A.Curtiss, F.Weinhold, Chem Rev, 88 (1988) 899.
- Glendening et al., E. D. Glendening, A. E. Reed, J. E. Carpenter, and F. Weinhold. Theoretical Chemistry Institute and Department of Chemistry, University of Wisconsin, Madison, Wisconsin 53706.
- Majid Monajjemi and Fatemeh Mollaamin ,J. Comput. Theor.Nanosci. 9, 2208-2214 (2012)
- F.Mollaamin,M.T.Baei,M.Monajjemi,R.Zhiani,B.Honarparvar, Russian Journal of Physical Chemistry A 82 (13), pp. 2354-2361,2008
- M. Monajjemi, H. Baheri, and F. Mollaamin, Journal of Structural Chemistry. 52(1) 54-59, 2011.
- M.Monajjemi,L.Mahdavian,F,Mollaamin, Bull Chem.Soc.Ethiop ,2008,22(2),1-10.
- F. Mollaamin, Z. Varmaghani , and M. Monajjemi, Physics and Chemistry of Liquids. 49(3), 2011, 318–336
- Majid Monajjemi, FatemehMollaamin , TaherehKarimkeshteh , J. Mex. Chem. Soc. 2005, 49(4), 336-340.
- M. Monajjemi , J. Najafpour & F. Mollaamin,Fullerenes, Nanotubes, and Carbon Nanostructures, 21: 213–232, 2013

This work is licensed under a Creative Commons Attribution 4.0 International License.
![]()
A New Edition of Web of Science
Journal Impact Factor
2022: 0.5
Five Year: 0.8
Journal is Indexed in

Cabells Whitelist
![]()


