Molecular Structural Studies of Captopril Drug, Using Thermal Analysis, mass Spectral Fragmentation and Semi- empirical MO- Calculations
H. M. Arafa
Chemistry Department, Faculty of Science, Tabuk University; P.O.Box 741 Tabuk 71491, Saudi Arabia
DOI : http://dx.doi.org/10.13005/ojc/300429
Article Received on :
Article Accepted on :
Article Published : 11 Dec 2014
In this work captopril an antihypertensive (KPL) drug, was investigated using thermal analysis (TA) measurements (TG-DTA) in comparison with electron impact (EI) mass spectral (MS) fragmentation at 70 eV. Semi-empirical molecular (MO) calculations, using PM3 method in the neutral and positively charged forms of the drug. These include molecular geometry, bond order, charge distribution, heats of formation and ionization energy. The behavior of the drug under drug TA decomposition, reveal a moderate stability up to 160Co before a completely decomposition in the range 160-240 Co. The initial decomposition is due to COOH + CH3 loss, followed by SH loss. On the other hand, the molecular ion can easily fragmented by CO2 loss followed by SH loss. This is the best-selected pathway comparable with decomposition using TA. MO-Calculation is used to declare these observations.
KEYWORDS:Captopril; thermal analysis; electron impact; MO calculation
Download this article as:| Copy the following to cite this article: Arafa H. M. Molecular Structural Studies of Captopril Drug, Using Thermal Analysis, mass Spectral Fragmentation and Semi- empirical MO- Calculations. Orient J Chem 2014;30(4). |
| Copy the following to cite this URL: Arafa H. M. Molecular Structural Studies of Captopril Drug, Using Thermal Analysis, mass Spectral Fragmentation and Semi- empirical MO- Calculations. Available from: http://www.orientjchem.org/?p=5774 |
Introduction
Captopril (KPL) (1-(3-mercapto-z-1)-methyl-1-oxopropyl)-l-proline (S,S) is a potential drug for the treatment of high blood pressure that can result from over production of angiotension II. Captopril inhibits the enzyme, which catalyzes the conversion of inactive angiotension II (schem-1) . The mechanism of the action has not been proved, but probably accurs through interaction with the enzyme a zinc containing metalloprptien, at its active site (1) . Captopril is an antihypertensive drug currenlly being administered in tablet from.
Several methods are in use for captopril analysis. These include spectrophotometry (2), chromatography (3), electro ( 4), AAS (5 ) .
Ford and timmins [6 ], pointed out use of thermal analysis in the characterization of pharmaceutical solids and the use of thermal analysis in the development of solid dosage forms.The decomposition rate of KPL was studied using isothermal and rising temperature methods on a simultaneous thermal analysis TG-DTA unit (7 ). The thermal behavior of KPL and its cabalt complex was studied ( 8) . Mass spectrometry is an indispensable tool in chemistry, biochemistry , pharmacy, and medicine (9 ).The technique is important for drug metabolism studies because it provides a large amount of structural information with little expenditure of sample (10 ). In electron impact ( EI ) mass spectrometry , the fragmentation consist of a series of competitive, and consecutive unimolecular fragmentation ( 11). Thermo gravimetric ( TG / DTG ) analysis is used to provide quantitative information on weight losses due to decomposition and evaporation of low molecular material as a function of time or temperature .
In conjunction between mass spectral fragmentation , and thermal decomposition process, the nature of the interpretation of thermal degradation can provide additional data, which can be used successfully for interpretation of experimental results of fragmentation processes (12 ) .
Although the literature is wealthy in information related to the biological activities of KPL, and its derivatives , it seems a lack of any correlation between chemical behavior, and its electronic structure of this drug.
In the present work, we have investigated the behavior of KPL, upon electron impact mass spectrometry at 70 eV, and thermal analysis (TA) measurement (TG-DTA, and DSC). Also MO calculation are performed using PM3 procedure on the neutral molecular and charged species to investigate bond order, atomic charge distribution, and heat of formation . These calculation are correlated with that obtained from both TA and ms to know information about the stability of the drug, and prediction of the site primary fragmentation step, and subsequent ones.
Experimental
Material
Sigma Chemical Co. applied the sample of captopril (Lot # 37H1120), St. Louis, Missouri.
Equipment and Procedures
Thermal Analyzer
A simultaneous TG-DTA Model No. 2960 (TA Instruments) was used to study the decomposition of the drug. The experiments were performed between ambient and 600 0C. The temperature program had a heating rate of 0C/ min from ambient to 600 0C.
Dry nitrogen, at a flow rate of 100 MI / min, was used as the purge gas. The flow rate was measured using an electronic flow meter, (J& W Scientific, Model # ADM 1000). The sample mass was kept in the range of 5mg. Platinum crucibles of 110 ml capacity were used as the sample holder and reference.
Mass Spectrometer
The mass spectrum of (KPL) was recorded with mass spectrometer Model MS-5988 GC Hewlett-Packard instrument operating at ionization voltage of 70 eV, and source temperature 200 C0, sample was evaporated from heated metal capillary and introduced to the source of mass spectrometer.
Quantum chemical Calculations
The calculations were performed using semi-empirical MO-calculation. The program used in these computations is the parametric PM3 method described by Stewart [13]. The geometries of all stable species studied were completely optimized with respect to all geometrical variables using the modified Davidson-Fletcher-Powell (DFP) [14] algorithm incorporated with the program. The program is run under the molecular orbital calculation package MOPAC 2000 by Stewart [13]for microcomputers and EF routine eigen vector following J. Baker method [15].
Results and discussion
The chemistry and reactivity of KPL drug have always been of grate interest because of its importance as an antihypertensive drug. Knowledge of thermal decomposition mechanism of KPL is very important to understand the chemical processes that exact major fragmentation pathway in EI using conventional MS. Combination of the experimental techniques (MS and TA) and semi-empirical MO calculation is very important to understand the following topics in TA.
*Stability of the drug in neutral and ionized species
*Primary site of fragmentation
*Major fragmentation pathway in TA and MS
*Selection of the most probable decomposition pathway of KPL
Thermal Analysis (TA)
From the TGA and DTA of captopril in an inert atmosphere of N2. KPL by itself decomposed over the temperature range 160-450 0C; a small residue of carbon was left at the end of the experiment. The experimental on the TG curve show two decomposition steps using a dry nitrogen atmosphere at a flow rat of 100 ml/min. Under the experimental conditions the onset weight loss temperature was around 160 0C, and followed by the second step at 290-310 0C from the TG curve readings. The first noticeable small blip on the DTG . curve is the peak temperature for the melting of KPL. The weight loss at 330 0C is assigned to SH loss or degradation.
The maximum signal (temperature) from the DTA curve provides a very sharp peak, and represents the melting point of the material tested. A shallow broad curve can also be disarmed. The melting point of the material causes the first sharp peak, and the dissociation of KPL in the region of the second shallow peak.
The first inflection occurs at 27% mass loss (160-240 0C) corresponding to the methyl, and carboxylic groups. The second step leads to the evaluation of the pyrrolinium groups as well as the partial pyrolysis of the molecule.
Fig. 3 reports the DSC of captopril. It can be seen that the figure displays an inverted endothermic through in the range 97-110 0C assigned to melting of the material as supported from DTA curve fig.1, with calculated heat enthalpy ∆H 110.403 J/g.
Mass Spectral Fragmentation of KPL
Electron ionization (EI) mass spectra of KPL drug at 70 eV was recorded and investigated. A typical mass spectrum of the drug (bar graph) is shown in Fig. 4, and the important ions are represented in table
Table- 1 R.I. for important ions produced from KLP drug
| m/z | R.Ia | Formula | Suggested Processes |
| 217 | 13.2 | [C9H15SNO3]+ | M+. |
| 199 | 6.3 | [C9H13SNO2]+ | [M-H2O]+ |
| 184 | 7.3 | [C9H14SNO3]+ | [M-SH]+ |
| 173 | 10.7 | [C9H14SNO]+ | [M-CO2]+ |
| 140 | 17.1 | [C8H14NO]+ | [M-SH-CO2]+ |
| 126 | 17.7 | [C7H12NO]+ | [M-CO2-CH2SH]+ |
| 98 | 4.6 | [C5H8NO]+ | [M-SH-CO2-C3H6]+ |
| 70 | 100% | [C4H8NO]+ | [M-SH-CO2-C3H6-CO]+ |
a:Relation to the base peak at 100%
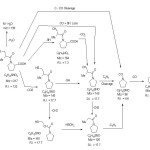
The main fragmentation pathways following electron impact of KPL drug may be rationalized to electron elimination of the molecular ion [M]+ as represented by(scheme2).
 |
Scheme 2 Click here to View scheme |
The most important mode of fragmentation of KPL drug molecular ion is that due to the competitive loss of CO2 and SH from M+. The signal appear at values m/z =173 [M-CO2] and at m/z = 173 [M-SH]+ confirm his assumption. On the other hand, the formation of the fragmentation at signal at m/z = 140 is due to the successive loss of CO2 and SH from molecular ion i.e. [M-CO2-SH] + or [M-SH-CO2]+ (C8H14NO+, R.I. 17.1%)
From Table (1) we can briefly summarized in the following main points
- No appreciable change on the change of the charge localization on the three O-atom upon ionization
- Higher change on the charge localized on the Sulpher (S) atom upon ionization (-0.030 to 0.798) which is surly sit of electron rupture.
- Moderate change of charge localized on nitrogen atom upon ionization
- Little difference in bond lengths between neutral molecule and charge species
- For bond order, the lowest values are at C2-C11 and C6-C7, we notice that for neutral C2-C1 < C6-C7 but for ion C2-C4 > C6-C7
Theoretical Calculations
MO calculations give valuable information about the structure of the molecules, which actually be used to support the experimental evidence. The much important parameters calculated using MO calculation include geometries, bond order bond length, charge distribution and heat of formation table(2). In the present work, the calculations have been carried out on captopril drug, neutral molecule and charged molecular ion and have been used for prediction of the weakest bond cleavage and follow the fragmentation pathway in neutral molecule (as in the TA decomposition) and charged molecular ion (as in the MS fragmentation).
Table(2) Bond order and charged species of (KPL) drug.
| Atom | Partial | Partial | Bond | Bond | Bond | Bond | Bond |
| Charge | Charge | Order | Order | Length | Length | ||
| (Neutral) | (Cation) | (Neutral) | (Cation | Ǻ(Neutral) | Ǻ(Neutral) | ||
| N1 | -0.060 | -0.025 | N1-C2 | 0.960 | 0.957 | 1.491 | 1.904 |
| C2 | -0.023 | -0.024 | C2-C3 | 0.972 | 0.980 | 1.537 | 1.535 |
| C3 | -0.102 | -0.100 | C3-C4 | 0.985 | 0.989 | 1.527 | 1.528 |
| C4 | -0.101 | -0.104 | C4-C5 | 0.991 | 0.997 | 1.523 | 1.527 |
| C5 | -0.072 | -0.068 | C5-N1 | 0.959 | 0.961 | 1.426 | 1.492 |
| C6 | 0.260 | 0.260 | N1-C6 | 1.046 | 1.134 | 1.426 | 1.409 |
| C7 | -0.083 | -0.085 | C6-C7 | 0.920 | 0.906 | 1.529 | 1.539 |
| C8 | -0.126 | -0.139 | C7-C8 | 0.984 | 0.991 | 1.529 | 1.522 |
| C9 | -0.162 | -0.277 | C7-C9 | 0.987 | 0.977 | 1.523 | 1.522 |
| S10 | -0.030 | 0.798 | C9-S10 | 0.992 | 1.030 | 1.523 | 1.532 |
| C11 | 0.375 | 0.370 | C2-C11 | 1.820 | 1.802 | 1.829 | 1.772 |
| O12 | -0.380 | -0.406 | C11-O12 | 1.049 | 1.089 | 1.526 | 1.527 |
| O13 | -0.306 | -0.280 | C11-O13 | 0.958 | 0.962 | 1.216 | 1.219 |
| H15 | 0.108 | 0.119 | C2-H15 | 0.981 | 986 | 1.355 | 1.347 |
| H16 | 0.061 | 0.076 | C3-H16 | 0.978 | 0.982 | 1.20 | 1.118 |
| H17 | 0.071 | 0.086 | C4-H17 | 0.980 | 0.985 | 1.107 | 1.107 |
| H18 | 0.062 | 0.072 | C4-H18 | 0.980 | 0.986 | 1.106 | 1.106 |
| H19 | 0.068 | 0.072 | C4-H19 | 0.966 | 0.979 | 1.05 | 1.106 |
| H20 | 0.054 | 0.057 | C5-H20 | 0.965 | 0.969 | 1.107 | 1.107 |
| H21 | 0.074 | 0.060 | C5-H21 | 1.804 | 1.789 | 1.113 | 1.114 |
| O22 | -0.366 | -0.370 | C6-O22 | 0.950 | 0.966 | 1.221 | 1.224 |
| H23 | 0.096 | 0.099 | C7-H23 | 0.978 | 0.976 | 1.128 | 1.121 |
| H24 | 0.056 | 0.092 | C8-H24 | 0.985 | 0.987 | 1.104 | 1.104 |
| H25 | 0.056 | 0.084 | C8-H25 | 0.983 | 0.986 | 1.098 | 1.099 |
| H26 | 0.064 | 0.042 | C8-H26 | 0.977 | 0.938 | 1.098 | 1.099 |
| H27 | 0.085 | 0.163 | C8-H27 | 0.976 | 0.919 | 1.105 | 1.118 |
| H28 | 0.090 | 0.166 | C9-H28 | 0.988 | 0.980 | 1.106 | 1.121 |
| H29 | 0.004 | 0.019 | S10-H29 | 0.988 | 0.980 | 1.308 | 1.298 |
| H30 | 0.227 | 0.239 | O13-H30 | 0.916 | 0.918 | 0.952 | 0.953 |
Fig. (5). Shows the numbering system of KPL skeleton that helps in ordering of charge distribution for neutral and charged (between brackets) and heat of formation. Bond order for both neutral molecule and charged molecular ion are presented in table 2.
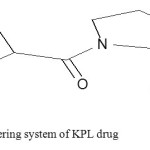 |
Fig4: The numbering system of KPL drug Click here to View figure |
MO- Calculations Versus Thermal Decomposition and Mass Spectral
Captopril is important drug used as antihypertensive. The scope of this investigation is restricted to search for predictions and decomposition of initial bond rupture during the course of fragmentation of the KPL molecule. The calculations are important to help in determination of first step of cleavage. Comparing the calculated data obtained from neutral molecule obtained from TA and that obtained from ionic species with that obtained from MS.
For neutral molecule, and on the base of MO- calculation the bond order side chain (Table 1) refers to the possible starting decomposition of the neutral molecule at C2-C11 (lowest bond order at 0.913) followed by rupture of bond C7-C8 (bond order = 0.984). This last bond is weakness due to electrostatic repulsion due to similar charge on C7 and C8 atoms. The rupture of these two bonds i.e. C2-C11 and C7-C8 loss of the COOH and CH3 molecules (wt loss= 27%). Subsequent decomposition the reminder loss 15.2% due to loss of sulohydryl radical (.SH), bond order C9-H10= 0.992. Such a behavior is not unexpected as S-C bond is weaker than C-C bond [16].The charge distribution on different atoms and heats of formation ∆HF (KJ/mol) for neutral and cation (KPL) equal -14.63, 62.3 respectively , the dipole moment of neutral and cation (KPL) 2.3 and 15.82 .
It is clear from the thermal analysis survey the KPL is stable up to 1580C and suffering from fast decomposition up to temperature range 3000C. This range contain different processes with very fast decomposition (low activation energy and low hilly life time m-1 this result refer to the high response of KPL drug to the temperature variation in the range 300-6000C .
The calculated charge density localized on different atoms (Table 1) refer to a high charge density on O12 (-0.380) followed by O22 (-0.366) followed by O13 (0.306) and low on N1 (0.060) and S10 (-0.03) while a high positive charge localized on C6 (0.260) due to adjacent to pair electron (i.e N and O), which may be form stabilizing remainder of the last stage.
Empirical observations indicate that the course of subsequent fragmentation is determined to large extent by the initial bond rupture of molecular ion in MS [17].
Mass spectra of KPL reveal two major competitive processes including principal fragmentation pathway (Scheme), due to loss of CO2 subsequent by SH or SH subsequent by CO2.
PM3 calculations data (table2) show that the C2-C11 has the smallest bond order of the charged system.
Finally we can proposed the thermal decomposition process as follow
schem2
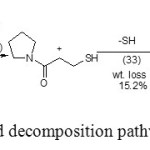 |
Scheme 2: The proposed decomposition pathway of(KPL) Click here to View scheme |
Conclusions
This study provides further insights into application of experimental TA and MS techniques and theoretical investigation using PM3 procedure on KPL drug. From the application of both practical and theoretical techniques it is concluded that: The primary fragmentation of KPL both excremental technique can be explained by COOH+CH3 loss followed by SH loss from the initial KPL molecule. In MS the first cleavage are due to the same initial bond cleavage while according with H- migration and loss of CO2 molecule. The MO- calculation data helps the selection of the most probable fragmentation pathway.
References
- M.A.Ondetti,B.Rubinand D.W.Cushman, Science, 1977, 196, 441.
- D.Temyu, M. Catala-lcardo and J. Martinez-calatyud, J.Pharm-Biomed Anal. 2004, 36(3), 549.
- T. Nishikaw , Y. Abe-Risudo , A. Yamada and K. Tahara, Analytical –Sciences, 2004, 20(10), 1395.
- W. Siangproh, P. Ngamukot, D. Chailapakul, Sensors and Actuators-B, 2003, 91(1-3), 60.
- M.A. El-Ries, F.A. Abou-Attia and I.M.M. Kenawy, J. Pharm-Biomed Anal. 2000, 23(2-3), 249.
- J.L Ford, P. Timmins, Pharmaceutical Thermal Analysis Techniques, and Applications, Willey, New York, 1989.
- Y. Huang, Y. Cheng, K. Alexander , D. Dollimore, Thermochemica Acta 2000, 6428, 1.
- R. Bucci, A.D. Magri, A.L. Magri, Thermochemica Acta, 1990, 161, 309.
- B. S. Larsen, C.N. MeEwen (Eds), Mass Spectrometry of Biological Materials, Marcel, Dekker, New York, 1998.
- K. Levesen, Fundamental Aspectes of Organic Mass Spectrometry, Verlag Chemie, Weinheim, New York,1978
- S. Bourcier, Hoppilliard, Eur. J. Mass Spectrom. 2003, 9, 35
- A. Somogi, A. Gomory, K. Vekey, Tamas, J.Org. Mass Spectrom. 1991, 26, 936
- J.I.P. Stewart, optimization parameters for semi-empirical methods I-methods, J. Comp. Chem. 1989, 209, 10
- J.I.P. Stewart, Fujitsu Limited, Tokyo, Japan, Efautine (Eigen Vector Following), 1999)
- J. Baker, Antilogarithm for the location of transition , J. Comp. Chem. 1986, 385.
- K. Levesen , Fundamental Aspects of Organic Mass Spectrometry, Verlag Chemie, Weiheim, New York, 1978.
- S. Bourcier, Hoppilliard Eur. J. Mass Spectrum 2003, 9, 35.

This work is licensed under a Creative Commons Attribution 4.0 International License.
![]()
A New Edition of Web of Science
Journal Impact Factor
2022: 0.5
Five Year: 0.8
Journal is Indexed in

Cabells Whitelist
![]()


