Flash preparation of carbenoids: A different performance of cyanogen bromide
Mohammad Jalilzadeh Hedayati1*, Nader Noroozi Pesyan2
1Department of Chemistry, Faculty of Science, Ahar branch, Islamic Azad University, Ahar, Iran.
2Department of Chemistry, Faculty of Science, Urmia University, Urmia, Iran.
DOI : http://dx.doi.org/10.13005/ojc/300477
Article Received on :
Article Accepted on :
Article Published : 19 Nov 2014
Cyanogen halides are known substances for the cyanating reaction. There are a few evidences for bromination reaction too. On the other hand carbenes are known as very important substances due to their remarkable reactions. Unfortunately carbenes at room temperature are very unstable and there is not a simple method for preparation of them. In most cases the isolation is not possible. We have reported a new reliable and fast preparation method of almost stable carbenoids. The mechanism of the formation has been discussed.
KEYWORDS:Carbene; Carbenoid; Cyanogen halides; Active carbons
Download this article as:| Copy the following to cite this article: Hedayati M. J, Pesyan N. N. Flash preparation of carbenoids: A different performance of cyanogen bromide. Orient J Chem 2014;30(4). |
| Copy the following to cite this URL: Hedayati M. J, Pesyan N. N. Flash preparation of carbenoids: A different performance of cyanogen bromide. Available from: http://www.orientjchem.org/?p=5466 |
INTRODUCTION
Cyanogen halides are known as important synthetic substances since they are strong cyanating agents. For instance cyanogen iodide reacts with the tertiary amine ring of marcfortine A to give cyano and iodocyano substituted products1. Also cyanogen iodide has been used as a reagent for disulfide bond formation in peptides2.
Cyanogen bromide is a very useful and extensively used reagent for the synthesis of cyanamides3. It is a colorless or white crystalline solid that decomposes in the presence of moisture and has a very small liquid range (mp 50–53 °C, bp 61–62 °C). It is cheap and can be synthesized by the reaction of sodium cyanide with bromine in aqueous medium4. CNBr produces electrophilic cyanide. Therefore, it is attacked by nucleophiles such as amines, alcohols and thiols.
Primary and secondary amines react with CNBr to give mono- and dialkylcyanamides5,6,7. CNBr is a very useful reagent for the preparation of aryl nitriles. It condenses with toluene in the presence of AlCl3 to give p-toluonitrile8. CNBr is known to convert carboxylic acids to nitriles at higher temperature under sealed-tube conditions. The reaction involves the intermediacy of a carboxylic cyanic anhydride. In anhydrous benzene, the carboxylic cyanic anhydride may be trapped and, in the presence of pyridine, symmetrical anhydrides may be produced in yields of 78–95%9. Von Braun found that a dialkyl thioether is cleaved by CNBr, at a somewhat elevated temperature (60–70 °C, sealed tube), to an alkyl thiocyanate and an alkyl bromide10. More recently, this reaction has been used for the selective cleavage of methionine peptides11 and other peptides12. Reaction between cyanogen bromide and some 3-amino-1,1-diphenylpropanes, variously substituted at C-1, leads either to N-cyano derivatives or to tetrahydrofurans13. Cyanogen bromide is a capable reagent for the synthesis of cyanamides 3, cyanates 14, and also used to selective cleavage of the methionyl peptide bonds in ribonuclease 15 and etc. Nonetheless this compound also is useful to simultaneity brominating and cyanating; the bromination and cyanation of imidazoles 16, free radical reaction with alkanes that result in bromination of alkanes 17 and α-bromination of β-aminoenones 18. By the way the Von Braun reaction is an organic reaction in which a tertiary amine reacts with cyanogen bromide to an organocyanamide19. The reaction of tertiary amines with CNBr yield disubstituted cyanamides and an alkyl bromide20. The bromodealkylation reaction has also been used to cleave the ring system of aziridine compounds21. Routine reactions of Cyanogen bromide have been presented in scheme 1. Although cyanogen bromide is a well known reagent, it has never been used as a carbenoid generator before.
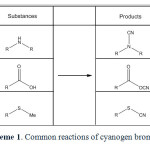 |
Scheme1: Common reactions of cyanogen bromide. Click here to View scheme |
RESULTS AND DISCUSSION
In this paper a new reaction of active carbons (1a-j) with cyanogen bromide in the presence of an organic base (triethyl amine or pyridine in some cases) has been described. Generated active carbenoids (2a-j) are shown in Scheme 2. Recently, we have performed the condensation of (thio)barbituric acids (1a-d) with aldehydes 22 and ketones 23 in the presence of BrCN and Et3N afforded spiro[furo[2,3-d]pyrimidine-6,5_-pyrimidine]pentaones and active carbenoid salt (2a-d). In the reaction of 5,5-dimethylcyclohexane-1,3-dione (dimedone, 1e) under same condition the similar scaffold structures have been produced as expected 24. Also we have investigated the condensation of Meldrum’s acid (1f) with aldehydes in the presence of BrCN and Et3N under the same condition. In this reaction, full-substituted cyclopropanes and the salt of 2f were obtained 25,26.
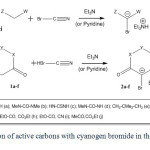 |
Scheme2: Reaction of active carbons with cyanogen bromide in the presence of TEA. Click here to View scheme |
On the other hand based on our earlier experiments, we decided to investigate on the possibility of the reaction of non-cyclic active carbons. For this reason some reactions of cyanogen bromide and triethyl amine with acyclic active carbons (1g-j) were performed 25,26. Although we did not attain triethylammonium 2-bromo-1,3-diethoxy-1,3-dioxopropan-2-ide (2h) obtained from diethyl malonate reaction (and other acyclic active carbon), we have succeeded in obtaining triethylammonium bromodicyanomethanide (2g) in the latter case malononitrile. Perhaps it is due to the stronger electron-attracting nature of cyanide group versus ester and/or simple carbonyl groups. By the way cyanide group has less steric hindrance than ester or carbonyl groups.
Proposed mechanism has been explained in Scheme 3. On the basis of the well established chemistry of pyrimidines27 and according to the mechanism of the bromination of 1-alkyl imidazoles by cyanogen bromide28 it is reasonable to assume that active carbons 1a-f reacted with cyanogen bromide and formed alkyl carbonobromidoimidate 3a-f. Intramolecular rearrangement of 3a-f afforded 4a-f followed by loss of HCN. Triethylamine as a base captured the acidic methylene proton of 4a-f and salts of 2a-f were formed. The salt of triethylammonium hydrobromide was also observed. Unfortunately, all attempts failed to separate or characterize 3a-f and 4a-f intermediates as representatives.
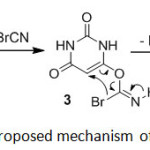 |
Scheme3: Proposed mechanism of 2a formation. Click here to View scheme |
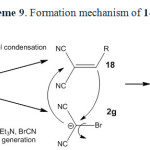
In the case of malononitrile a similar mechanism has been suggested (scheme 4).
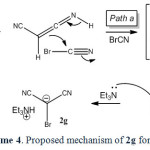 |
Scheme4: Proposed mechanism of 2g formation. Click here to View scheme |
It is reasonable to assume that the compound 1g reacts directly with cyanogen bromide to form 2,2-dicyanoacetimidoyl bromide (5) intermediate through path a. Intramolecular rearrangement of this intermediate produces 2-bromomalononitrile (6) intermediate. Finally, triethylamine as a base captures the acidic proton affording 2g. Malononitrile (1g) directly reacts with triethylamine to form the salt of triethylammonium dicyanomethanide in the absence of cyanogen bromide (path b). The salt of triethylammonium dicyanomethanide was also separated and characterized in direct reaction of 1g with triethylamine through path b.
All attempts to isolation of 5 and 6 were unsuccessful unfortunately. According to our search, there is no report about compound 2g in literatures. The salt of 2g was isolated and characterized by spectroscopy techniques. Other evidence for the formation and confirmation of 2g (the existence of bromine atom in this salt structure) was performed by Beilstein test and the wet silver nitrate test (precipitate of pale yellow silver bromide).
The IR spectrum of 2g shows the frequency of NH+ stretching at the broad range of 2500-3300 cm-1, frequency of CN and C-Br stretching at 2167 and 565 cm-1, respectively. The 1HNMR spectrum of this compound shows (integration in parenthesis) a triplet at δ 1.39 ppm (9H) and a multiplet (a quartet approximately) at 3.14 ppm (6H) are corresponding to methyl and methylene protons in triethylammonium salt moiety, respectively. A broad singlet at δ 9.78 ppm (1H) is of NH+ in this salt. 13C NMR spectrum of this salt shows four distinct peaks that confirm the structure of 2g.
The structure of the 2a-f was deduced from their IR, 1H NMR, 13C NMR spectra and CHN analysis. Representatively, the 1H NMR spectrum of 2a consisted of a triplet for three methyl protons at δ 1.16 ppm, a quartet for the corresponding methylene protons at 3.08 ppm, a broad singlet for ammonium proton at δ 8.93 ppm and a singlet for two amide protons at δ 9.38 ppm on barbituric acid ring moiety. The IR spectrum of 2a indicated no CN bond stretching absorption. The 13C NMR spectrum of 2a showed five distinct peaks. The peak at δ 9.09 and 46.19 ppm are of methyl and methylene carbons in triethylammonium salt moiety, respectively and the peak at 72.27 ppm corresponds to C-5 on barbituric acid ring. Two peaks at δ 152.02 and 161.26 ppm correspond to the carbonyl groups on barbituric acid ring moiety, respectively. Other evidence for the formation of 2a-f (the existence of bromine atom in these molecules) was performed by Beilstein test and the wet silver nitrate test29 (precipitate of pale yellow silver bromide). During isolation and identification of 2a-f interestingly there is not any evidence of cyanide peak in IR spectra (scheme 5).
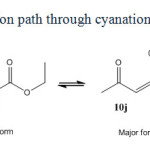 |
Scheme5: Reaction path through cyanation of active carbon. |
In the case of Barbituric acid’s salts, 2b plays an important role and its nucleophilicity is stronger than that of 2a and 2c. The 2a and 2c have aromatic nature with delocalizing of the negative charge while 2b has not.
Acyclic compounds with the exception of malononitrile behave in different way. Reason of this different behavior is not clearly recognized. Perhaps it is due to the enol-keto equilibrium occurred in acyclic beta-dicarbonyl or beta-cyanocarbonyl compounds (scheme 6). For example ethyl aceto acetate mainly exists in beta-keto 1-en 1-ol (12j) form and not in beta dicarbonyl (1j) form. Whereas cyclic compounds like Barbituric acid, Dimedone and Meldrum’s acid (1a-f) have been founded mainly in keto form. A similar equilibrium is not seen in malononitrile (scheme 7). Bromodicyanomethanide (2g) is the main form. Any symptom of other resonance forms is not seen in 13CNMR spectra of 2g as expected. Also as seen in figure 2, three distinctive peaks for 2a and two peaks for ammonium ion are shown in 13CNMR spectrum. There is not any symptom of existence of enol form for 2a.
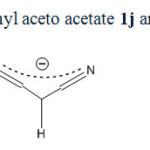 |
Scheme6: Resonance forms in ethyl aceto acetate 1j and 1-ethoxy-1,3-dioxobutan-2-ide (9j)Click here to View scheme |
Reaction of diethyl malonate (DEM) (1h), ethyl cyanoacetate (ECA) (1i) and ethyl acetoacetate (EAA) (1j) with cyanogen bromide in acetone formed diethyl bromomalonate (13h) and ethyl bromoacetoacetate (13j) in moderate yield, respectively. Triethylammonium hydrobromide salt was observed in all reaction products (scheme 8).
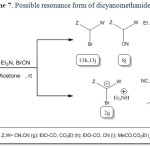 |
Scheme7: Possible resonance form of dicyanomethanide (2g). Click here to View scheme |
The 1H NMR spectrum of diethyl bromomalonate, 13h show a triplet at δ 1.31 and a quartet at δ 4.29 ppm are of to equivalent methyl and methylene protons, respectively. A singlet at δ 4.82 corresponds to the CHBr proton. The peak integration ratios 1.0:4.0:6.0 are of methine, two methylene and two methyl protons, respectively. These ratios are in good agreement with the structure of 13h. The 13C NMR spectrum of 13h show a peak at δ 13.85 and 63.21 ppm are of methyl and methylene carbon atoms, at δ 42.40 and 164.58 ppm corresponds to the methine and carbonyl carbon atoms respectively. Diethyl cyanomalonate, 8h has been synthesized previously by the reaction of ethyl cyanoacetate, 1i with ethyl chloroformate in acetone30. We performed the reaction of 1h with cyanogen bromide according to method of reference 30 (formation of 8h was not observed).
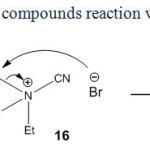 |
Scheme8: Acyclic compounds reaction with cyanogen bromide. Click here to View scheme |
In contrast, our experiments indicated that the reaction of 1h with cyanogen bromide leads to diethyl bromomalonate, 13h and N,N-diethylcyanamide (14) in acetone (Scheme 9).
 |
Scheme9: Formation mechanism of 14. Click here to View scheme |
Expectedly, triethylamine attacks the NC+ of cyanogen bromide obtaining triethylcyanoammonium bromide salt (16) (Scheme 9). According to Scheme 9, the ethyl bromide and 14 were observed based on von Braun reaction through analyzing the crude reaction mixture by GC-Mass analyses. The reaction of ethyl cyanoacetate, 1i with cyanogen bromide in ether indicated no significant product(s). In contrast, in acetone as a polar aprotic solvent ethyl 2-isopropylidenecyanoacetate (15i) was afforded by means of the condensation of 1i with acetone in 32% yield (Scheme 8). No 13i and 8i were observed in this reaction.
The structure of 15i was characterized by 1HNMR, 13CNMR and GC-Mass analysis. Interestingly, ethyl bromoacetoacetate, 13j or ethyl cyanoacetoacetate, 8j were found in the reaction of ethyl acetoacetate, 1j and cyanogen bromide in ether as solvent (Scheme 8).
For assurance of existence of free carbenoid, in the case of malononitrile we have performed some of well-known carbenes reactions. Entrapping of free carbenes by polar carbon-carbon double bonds (producing cyclopropane structure) is one of the most famous reactions used to identification of carbenes. Entrapping includes the shuttle attack of carbene to polar carbon-carbon double bond. Two moles of malenonitrie were reacted with aldehydes 17k-z as knovenagel condensation (insitu performing of 18k-z). On the other hand malononitrile was reacted with trithylamine and cyanogen bromide as described before. Negative charge of 2g attacks to polar double bond of 18k-z as shown in the scheme 10. Final cyclopropane products 19k-z were isolated and identified by 1HNMR, 13CNMR and IR data. Existence of free carbene was maintained by results. The same method for entrapping of carbenes in the case of cyclic substrates resulted similar cyclopropanes as expected.
 |
Scheme10: Insitu generation of carbene based on malononitrile |
EXPERIMENTAL
General procedures
Melting points were measured with a digital melting point apparatus (Electrothermal) and were uncorrected. IR spectra were determined in the region 4000- 400 cm-1 on a NEXUS 670 FT IR spectrometer by preparing KBr pellets. The 1HNMR and 13CNMR spectra were recorded on Bruker 300 FT-NMR at 300 and 75 MHz, respectively (Urmia University, Urmia, Iran). 1HNMR and 13CNMR spectra were obtained on solution in DMSO-d6 and/or in CDCl3 as solvents using TMS as internal standard. The data are reported as (s=singlet, d=doublet, t=triplet, q=quartet, m=multiplet or unresolved, bs=broad singlet, coupling constant(s) in Hz, integration). All reactions were monitored by TLC with silica gel-coated plates (solvent system of most cases is AcOEt:AcOH/ 80:20/ v:v). Elemental analyses were performed using a VarioEL III analyzer (Tabriz University, Tabriz, Iran). The mass analysis performed using mass spectrometer (Agilent Technology (HP) type, MS Model: 5973 ,ion source temperature was 230 ºC (Tehran University, Tehran, Iran).
Synthesis of 2, Method A: In a 25 mL round bottom flask equipped by a magnetically stirrer, dissolved 0.12 g (0.96 mmol) cyanogen bromide (BrCN) in 4 mL ethanol at 0 °C. Then separately, 0.15 g (0.96 mmol) barbituric acid were dissolved in 10 mL ethanol in an Erlenmeyer, 0.75 mL triethylamine was added into solution and then was transferred it into a separatory funnel, then it was added drop wise into solution of BrCN in round bottom flask at 0 °C. The progression of reaction was monitored by thin layer chromatography (TLC). After a few minutes, the white-cream solid precipitate, filtered off, washed with few mL ethanol and dried.
Method B: In a 10 mL with Teflon-faced screw cap tube equipped by a magnetically stirrer, dissolved, 0.132 g (2 mmol) malononitrile, 0.202 g (2 mmol) triethylamine in 5 mL ethanol and then 0.210 g (2 mmol) cyanogen bromide (BrCN) was added into solution at 0 °C. White-Cream color solid precipitate in less than five minutes, filtered off and washed with few mL ethanol and dried.
Synthesis of cyclopropane structures: In a 10 mL with Teflon-faced screw cap tube equipped by a magnetically stirrer, dissolved, 0.265 g (4 mmol) malononitrile, 0.400 g (4 mmol) triethylamine and 2 mmol aldehydes in 7mL ethanol and then 0.210 g (2 mmol) cyanogen bromide (BrCN) was added into solution at 0 °C. temprator was raised to 70 °C. Cream color solid precipitate after a few minutes, filtered off and washed with few ml ethanol and dried.
The physical and spectral data of the selected compounds are follows as representatives. Spectral data for intermediate compounds (which prepared for study of mechanism) have not been described. Do not hesitate to ask for full supplementary data or more information.
Triethylammonium-5-bromo-2,4,6-trioxohexahydropyrimidin-5-ide (2a)
White solid (50%); mp = 155-158 °C (decomps.); FT-IR (KBr) 3130, 2985, 2816, 1658 (C=O), 603, 524 (C-Br) cm-1; 1H NMR (DMSO-d6, 300 MHz) δ 1.16 (t, 9H, 3CH3), 3.08 (q, 6H, 3CH2), 8.93 (bs, 1H, NH ammonium), 9.38 (s, 2H, 2NH BA); 13C NMR (DMSO-d6, 75 MHz) δ 161.3, 152.0, 72.3, 46.2, 9.1; Anal. Calcd. for C10H18N3O3Br: C, 38.9; N, 13.63; H, 5.84. Found: C, 39.04; N, 13.66; H, 5.92 %. MS, m/z 308 (M+, 0), 154 (7), 128 (base peak, 100), 101 (15), 86 (60), 72 (5), 58 (16), 42 (98).
Triethylammonium-1,3-dimethyl-5-bromo-2,4,6-trioxohexahydropyrimidin-5-ide (2b)
White solid (45%); mp 152-154 °C (decamps.). FT-IR (KBr) 3200, 2980, 1670, 526 cm-1; 1H NMR (DMSO-d6, 300 MHz) δ 1.17 (t, 9H), 2.80 (s, 6H), 3.08 (q, 6H), 8.95 (bs, 1H); 13C NMR (DMSO-d6, 75 MHz) δ 161.9, 157.1, 73.4, 46.1, 30.2, 9.1; Anal. Calcd. for C12H22N3O3Br: C, 42.87; N, 12.50; H, 6.60. Found: C, 42.65; N, 12.43; H, 6.52%.
Triethylammonium-5-bromo-4,6-dioxo-2-thioxohexahydropyrimidin-5-ide (2c)
White solid (50%); mp = 159-161 °C; FT-IR (KBr) 3408, 3070, 2975, 2937, 2677, 1648 (C=O), 1612, 590, 526 (C-Br) cm-1; 1H NMR (DMSO-d6,300 MHz) δ 1.16 (t, 9H, 3CH3), 3.07 (q, 6H, 3CH2), 10.17 (bs, 1H, NH ammonium), 10.35 (bs, 2H, 2NH TBA); 13C NMR (DMSO-d6, 75 MHz) δ 174.7, 164.3, 79.7, 46.2, 9.1; Anal. Calcd. for C10H18N3O2SBr: C, 37.05; N, 12.97; H, 5.56. Found: C, 37.10; N, 13.05; H, 5.51 %.
Triethylammonium-2-bromo-5,5-dimethylcyclohexane-1,3-dione-2-ide (2e)
Colorless crystalline solid (62%); mp= 75-76 °C; FTIR (KBr, Cm-1) vmax: 3417, 2958, 2870, 2678, 2495, 1644, 1610, 1509, 472; 1H NMR (CDCl3, 300 MHz) δ 7.27 (bs, 1H), 3.02 (q, J=7.5 Hz, 6H), 2.18 (s, 4H), 1.17 (t, J=7.5 Hz, 9H), 0.87 (s, 6H); 13C NMR (CDCl3, 75 MHz) δ 8.7, 28.3, 31.7, 46.2, 50.2, 96.7, 186.0.
Triethylammonium 5-bromo-2,2-dimethyl-4,6-dioxo-1,3- dioxan-5-ide (2f)
White crystalline solid (45%), mp 252–253 °C; IR (KBr) 3448 (OH), 2995, 2944, 2738, 2678 (CH-aliph.), 1752 (C=O), 1033 (C–O), 752 (C–Br) cm-1; 1H NMR (CDCl3, 300 MHz) δ 10.10 (bs), 3.09 (q, 6H), 1.54 (s, 6H), 1.27 (s, 9H); 13C NMR (CDCl3, 75 MHz) δ 164.0, 102.2, 46.4, 27.5, 25.6, 8.7.
Triethylammonium bromodicyanomethanide (2g)
White crystalline solid (60); FT-IR (KBr) 3420, 3047, 2987, 2947, 2804, 2738, 2678, 2491, 2167, 1475, 1035, 565 cm-1; 1H NMR (CDCl3, 300 MHz) δ 1.39 (t, 9H, J = 7.2 Hz), 3.14 (m, 6H), 9.78 (bs, 1H); 13C NMR (CDCl3, 75 MHz) δ 141.6, 121.6, 46.6, 8.8.
Diethyl bromomalonate (13h)
The experimental procedure for the reaction of 1h, 1i and 1j with cyanogen bromide and triethylamine in acetone and other solvents were similar to the barbituric acid experimental procedure (method A). Colorless oily liquid (38%). FT-IR (KBr) 2985, 1743, 1150 cm-1; 1H NMR (DMSO-d6, 300 MHz): δ 1.31 (t, 6H), 4.29 (q, 4H), 4.82 (s, 1H). 13C NMR (DMSO-d6,, 75 MHz); δ 164.6, 63.2, 42.4, 13.9; GCMS m/z: (RT, 3.96 min.), 241 (M+2), 239 (M), 212 (2), 193 (10), 166 (18), 140 (30), 138 (42), 120 (20), 29 (100, base peak).
3-phenylcyclopropane-1,1,2,2-tetracarbonitrile (19k)
White crystalline solid (65%), mp 225-227 °C (decompds.) (lit. 229-230 °C10); FT-IR (KBr) 3046, 3013, 2928, 2264, 1640, 1374, 1231, 741, 701, 662 cm-1; 1H NMR (DMSO-d6, 300 MHz) δ 5.29 (s, 1H), 7.47 (m, 3H), 7.78 (m, 2H); 13C NMR (DMSO-d6, 75 MHz) δ 130.4, 130.0, 129.4, 127.3, 111.4, 109.9, 42.2, 23.6.
3-(2-nitrophenyl)cyclopropane-1,1,2,2-tetracarbonitrile (19l)
White crystalline solid (60%), mp 215-216 °C (decompds.); FT-IR (KBr) 3108, 3089, 3031, 2924, 2866, 2258, 1610, 1530, 1346, 851, 794, 736, 702 cm-1; 1H NMR (Acetone-d6, 300 MHz) δ 5.45 (s, 1H), 7.96 (t, 1H, J = 7.5 Hz), 8.07 (t, 1H, J = 7.5 Hz), 8.37 (d, 1H, J = 7.5 Hz), 8.51 (d, 1H, J = 7.5 Hz); 13C NMR (Acetone-d6, 75 MHz) δ 147.9, 135.3, 132.7, 132.6, 126.6, 122.0, 110.2, 108.8, 41.7, 23.8.
3-(3,4,5-Trimethoxyphenyl)cyclopropane-1,1,2,2-tetracarbonitrile (19p)
White crystalline solid (68%), mp 227-229 °C (decompds.); FT-IR (KBr) 3047, 2997, 2943, 2842, 2260, 1592, 1512, 1468, 1421, 1251, 1127, 993 cm-1; 1H NMR (Acetone-d6, 300 MHz) δ 3.77 (s, 3H), 3.87 (s, 6H), 4.96 (s, 1H), 7.31 (s, 2H); 13C NMR (Acetone- d6, 75MHz) δ 153.9, 139.8, 121.4, 110.7, 109.1, 107.2, 59.8, 55.9, 42.9, 22.9.
ACKNOWLEDGEMENTS
The Authors gratefully acknowledge financial support by the Research Council of Islamic Azad University-Ahar Branch.
REFRENCES
- 10. Lee, B. H.; Clothier, M. F.; Pickering, D. A. Tetrahedron lett. 1997, 38, 6119-6122.
- Bishop P., Chmielewski J. Tetrahedron lett. 1992, 33, 6263–6266.
- Kumar, V. Synlett. 2005, 10, 1638-1639.
- Hartmann, W. W.; Dreger, E. E. Org. Syn. Coll. Vol. 2; John Wiley: London, (1943).
- (a) Podesva, C. P.; Tarlton, E. J.; McKay, A. P. Can. J. Chem. 1962, 40, 1403-1407. (b) Deaton, D. N.; Hassell, A. M.; McFadyen, R. B.; et al. Bioorg. Med. Chem. Lett. 2005, 15, 1815-1819.
- Snider, B. B.; O’Hare, S. M. Tetrahedron Lett. 2001, 42, 2455-2458.
- Cai, T.; Xian, M.; Wang, P. G. Bioorg. Med. Chem. Lett. 2002, 12, 1507-1510.
- Scholl, R.; Kacer, F. Ber. Dtsch. Chem. Ges. 1903, 36, 322-331.
- Ho, J.-L.; Wong, C. M. Synth. Commun. 1973, 3, 63-66.
- von Braun, J.; Engelbertz, P. Ber. Dtsch. Chem. Ges. 1923, 56, 1573-1577.
- (a) Gross, E.; Witkop, B. J. Am. Chem. Soc. 1961, 83, 1510-1511. (b) Stadtmana, E. R.; Van Remmen, H.; Richardson, A.; et al. Biochim. Biophys. Acta 2005, 1703, 135-140.
- Zhong, H.; Marcus, S. L.; Li, L. J. Am. Soc. Mass Spectrom. 2005, 16, 471-481.
- Casy A.F., Hassan M.M.A., Tetrahedron let. 1967, 23, 4075–4086.
- Martin, D.; Bauer, M. Org Synth Coll. Vol. 7, p435. (1990)
- Gross, E.; Witkop, B. J. Am. Chem. Soc. 1961, 83, 1510-1511.
- McCallum, PBW.; Grimmett, MR.; Blackman, AG. Aust. J. Chem. 1999, 52, 159-166.
- Tanner, DD.; Lycan, G.; Bunce, NJ. Can. J. Chem. 1970, 48, 1492-1497.
- Alberola, A.; Andres, C.; Ortega, AG.; Pedrosa, R.; Vicente, M. Synth Commun.1986, 16, 1161-1165.
- Organic Syntheses, Coll. Vol. 3, p.608 (1955); Vol. 27, (1947).
- Hageman, H. A. The von Braun Reactions, In Organic Reactions, Vol. 7; Blatt, A. H.; Cope, A. C.; McGrew, F. C.; Niemann, C.; Snyder, A., Eds.; Wiley: New York, (1953).
- Furuya, S.; Okamoto, T. Heterocycles. 1988, 27, 2609-2618.
- Jalilzadeh, M.; Noroozi Pesyan, N.; Rezaee, F.; Rastgar, S.; Hosseini, Y.; Şahin, E. Mol. Div. 2011, 15, 721-731.
- Hosseini, Y.; Rastgar, S.; Heren, Z.; Büyükgüngör, O.; Noroozi Pesyan, N. J. Chin. Chem. Soc. 2011, 58, 309-318.
- Noroozi Pesyan, N.; Shokr, A. R.; Behroozi, M.; Şahin, E. J. of the Iranian Chem. Soc. 2013, 10, 565-575.
- Noroozi Pesyan, N.; Garib, A.; Behroozi, M.; Shokr, A. Arab. J. Chem. 2013, in press.
- Noroozi Pesyan, N.; Kimia, M. A.; Jalilzadeh, M.; Şahin, E. A. J. Chin. Chem. Soc. 2013, 60, 35-44.
- Brown, DJ.; Mason, SF. Chemistry of Heterocyclic Compounds. Vol. 16, John Wiley & Sons Inc. New York, (1962)
- McCallum, PBW.; Grimmett, MR; Blackman, AG.; Weavers, RT. Aust. J. Chem. 1999, 52,159-166.
- Schriner RL, Fusan RC, Curtin DY, Morrill TC The systematic identification of organic compounds, 6th, Edn, John Wiley & Sons, New York, (1980).
- Witherell, R. D. The Stereoselective Synthesis of 13C2-labelled Lysines. M. Sc. Thesis, Dalhousie University, Halifax, Nova Scotia, Canada, (1999).

This work is licensed under a Creative Commons Attribution 4.0 International License.
![]()
A New Edition of Web of Science
Journal Impact Factor
2022: 0.5
Five Year: 0.8
Journal is Indexed in

Cabells Whitelist
![]()


