Structure-Based Design of Inhibitors Against Maltosyltransferase Glge
Junie B. Billones*1,2 and Ariane Marielle F. Valle1
1Department of Physical Sciences and Mathematics, College of Arts and Sciences, Manila 1000, Philippines 2Institute of Pharmaceutical Sciences, National Institutes of Health, University of the Philippines Manila, Taft Avenue, Ermita, Manila 1000, Philippines.
DOI : http://dx.doi.org/10.13005/ojc/300326
Article Received on :
Article Accepted on :
Article Published : 12 Sep 2014
The emergence of Mycobacterium tuberculosis(Mtb) drug resistant strains calls for research of novel anti-TB drugs. In this study,structure-based pharmacophore generation, virtual screening, molecular docking and de novolead optimization were employed in the search for possible inhibitors of MaltosyltransferaseGlgE enzyme,a recently validated anti-TB drug target. Chemical libraries containing only natural products were screened. The top hits were docked and the resulting leads were subsequently modified usingDe Novo Evolution. Three modified compounds were found to have greaterbinding energy than the substrate. All were derived from the lead compound ZINC39010596 (5,7-dihydroxy-2-propan-2-yl-8-[(2S,3R,4S,5S,6R)-3,4,5-trihydroxy-6-(hydroxymethyl)tetrahydro-2H-pyran-2-yl]oxychromen-4-one). Toxicity predictions of these compounds revealed that they were non-carcinogenic, non-mutagenic, and biodegradable.
KEYWORDS:Tuberculosis; MaltosyltransferaseGlgE; Structure-based Pharmacophore Generation; Virtual Screening; Molecular Docking
Download this article as:| Copy the following to cite this article: Billones J. B, Valle A. M. F. Structure-Based Design of Inhibitors Against Maltosyltransferase Glge. Orient J Chem 2014;30(3). |
| Copy the following to cite this URL: Billones J. B, Valle A. M. F. Structure-Based Design of Inhibitors Against Maltosyltransferase Glge. Orient J Chem 2014;30(3). Available from: http://www.orientjchem.org/?p=4592 |
INTRODUCTION
Tuberculosis (TB) is a highly contagious airborne disease brought by the pathogen Mycobacterium tuberculosis (Mtb) that causes infection in the lungs1. Despite the fact that the causative organism was discovered 100 years ago and drugs against this agent are already available, TB remains to be a worldwide health problem2. It is considered by the WHO as one of the leading causes of death,affecting approximately 8.7 million people in 2011 alone, 1.4 million of which have died3. Management of TB is usually done using antibacterial drugs like isoniazid and rifampicin4. However, the emergence of multi-drug resistant TB (MDR-TB) and extensively drug resistant TB (XDR-TB) strains have now made these drugs less effective in killing the mycobacterium5. Thus, there is great need to develop novel anti-TB drugs.
One recently validated drug target for Mtb is the enzyme MaltosyltransferaseGlgE (Rv1327c)6. It is part of a previously unrecognized four step α-glucan pathway that converts the disaccharide trehaloseinto α-glucan polysaccharide which is a component of the extracellular α-glucan capsule of Mtb, important for its virulence and persistence.By employing traditional and chemical reverse genetics, GlgE inactivation caused rapid death of Mtb in vitro in mice by two mechanisms. The first is called “self-poisoning”; in which accumulation of the substrate ofMaltosyltransferaseGlgE, particularly maltose-1-phosphate (M1P), proved fatal to the bacterium because it elicits pleiotropic stresses which includes inhibition of respiration, induction of stringent response and DNA damage. Interestingly, Mtb responds to the stress by generating more trehalose, the M1P precursor, in a misguided stress protection response that further fuels the accumulation of M1P, worsening the case scenario.The second but independent death mechanism is based on the fact that the GlgE pathway works side-by-side with the Rv3032 pathway. As long as the redundant Rv3032 is functioning, the GlgE pathway could be responsible as a whole for the viability and virulence of Mtb. However, simultaneousinhibition of both pathways is lethal. This implies that while mono therapy with GlgE inhibitors is sufficient to kill Mtb as effective as first-line drugs like isoniazid, it could be further improved by combination therapy with Rv3032 inhibitors and could also avoid the possibility of development of resistance.This makes Maltosyl transfer as eGlgE a promising anti-TB drug target. Furthermore, it is absent in humans, but is present in almost all mycobacteria and other opportunistic pathogens like Pseudomonas and Burkholderia species. The structure of its homologue from Streptomyces coelicolor was released only in 2011 and was reported to have similar catalytic properties with MtbGlgEtogether with a conserved active site7. With an available structure of the drug target, structure-based drug design may now be employed.
In this work, structure-based screening and in silico optimization8 was carried out to search for potential inhibitors of MtbGlgE.Specifically, we sought natural products that possess significant structural and chemical complementarities to the target receptor9. Accordingly, a pharmacophore10 based on the binding site in GlgE wasgenerated and used to screen several compounds databases of natural products. The top hits were docked to the drug target and were rank-ordered based on their binding energy. The high-binding compounds were further optimized structurally. Lastly, the variants of the leads were evaluated for their potential toxicity properties.
MATERIALS AND METHODS
All computational procedures were done on Accelrys Discovery Studio (DS) Client v2.5.0.9164 installed on a computer running on Microsoft® Windows 7 Home Premium 64-bit Operating System using a processor of Intel® Core™ i3 CPU @ 2.27 GHz and installed memory (RAM) of 4.00 GB.
Retrieval of Maltosyl transferaseGlgE structure and compound libraries
Because the structure of Maltosyl transfer as eGlge from Mtbis not yet available, the crystal data of its closest homologue from another actinomycete, Streptomyces coelicolor, was used in this study. The 2.09 angstroms resolution 3D structure of Maltosyl transfer as eGlgE Is of orm I in complex with the lig and maltose (PDB code: 3ZT5) was retrieved from the RCSB Protein Data Bank.Only chain A among the four identical subunits (A, B, C and D)of the protein was retained. The protein was cleaned, prepared and minimized using the Clean Protein tool,Prepare ProteinandMinimization protocol. To determine the extent of modification that occurred on the protein due to the application of the said protocols, the RMSD value was computed using the Align and Superimpose Proteins protocol.
Natural products catalogs were obtained from ZINC database as SDF files (http://zinc.docking.org/browse/catalogs/natural-products), namely; Ambinter Natural Products, AnalytiCon Discovery NP, IB Screen NP,Indo fine Natural Products, Molecular Diversity Preservation International, Nubbe Natural Products, Princeton NP, Selleck Biochemicals NP, Specs Natural Products, TCM Database @ Taiwan and UEFS Natural Products. In addition to these, other compound libraries, particularly “MEGx: Pure Natural Products” and “NATx: Semi-Synthetic Screening Compounds based on Natural Product Scaffolds” were obtained from AnalytiCon Discovery (http://www.ac-discovery.com/content/Products&Technologies) while Otava Drug-like Green Collection was retrieved from Otava Chemicals (http://www.otavachemicals.com/download-compound-libraries/doc_details/5-drug-like-green-collection-sdf). Lastly, the ligand maltose was obtained as SDF from the PDB. The compounds were then prepared using the Prepare Ligands protocol and built into separate databases using the Build 3D Database protocol.
Structure-based Pharmacophore Generation
The bound maltose was used to locate the active site of the enzyme. A site sphere was generated on the active site using the Define Sphere from Selection tool. Site sphere optimization was done by adjusting the size of the sphere until the docked pose is as close as possible to the pose of the original crystallized structure. Afterwards, maltose was deleted and the protein was defined as the receptor using the Define Selected Molecule as Receptor tool.The active site was then analyzed for donor, acceptor, and hydrophobic features by generating a pharmacophore using the Interaction Generation protocol. The initial pharmacophore features were further trimmed using the Cluster Current Feature and Keep Only Cluster Centers tools for all acceptor, donor, and hydrophobe features.
Virtual screening and molecular docking
The above mentioned databases of molecules were then screened using the pharmacophore generated earlier by running the Screen Library protocol using rigid fitting method. Compounds with fit value greater than or equal to 3.0 were subjected to second screening, this time using the flexible fitting method. From here, compounds with fit value greater than or equal to 4.0 were subjected to molecular docking. Maltose was rescreened regardless of the fit value.
Molecular docking was done using the Dock Ligands (CDOCKER) protocol. The poses generated were evaluated by calculating their binding energies using the Calculate Binding Energies protocolwith In Situ Ligand Minimization and then ranked accordingly. Moreover, visual inspection of the docked poses was also conducted by generating ligand interaction diagrams. From this, the lead compounds were identified.
Chemical Modification (De Novo) of the Lead Compounds
The structures of the chosen lead compounds were modified using the De Novo Evolution protocol, Full Evolution mode. For this, the highest ranking pose of the lead compound was used as the ligand scaffold. The fragment library used was the built-in small fragment library, organic.str.The binding energies of the generated modified compounds were calculated and ranked accordingly, while ligand interaction diagrams were also generated and further analyzed for molecular interactions.
Toxicity Prediction of Modified Lead Compounds
The modified lead compounds were further tested for properties like carcinogenicity, mutagenicity and biodegradability using the Toxicity Prediction (Extensible) protocol.
RESULTS AND DISCUSSION
The structure of Maltosyl transfer as eGlgE (PDB code: 3ZT5) was first subjected to the Prepare Protein protocol to address certain issues11 like missing atoms, missing loop regions, etc. in order to standardize the protein for subsequent computational processes. Minimization was done to minimize the energy of the protein through geometry optimization. These protocols may bring about conformational changes; thus it is made sure that the extent of modification that occurred is well within the allowable range. In docking simulations, the calculated RMSD between the main chain atoms must be below 1.5 Å12. In this study, the computed RMSD was 0.721 Å (Fig. 1) which is well within the acceptable range, thus validating the prepared protein.On the other hand, the compound databases were subjected to the Prepare Ligands protocol to generate reasonable starting ligand structures and in order to apply the Lipinski filter. The Lipinski filter is a collection of criteria (H-bond donors < 5, H-bond acceptor < 10, Molecular Mass < 500, log P < 5) that are commonly observed in drug-like candidates13. Subsequently, the prepared ligands were built into separate 3D databases.
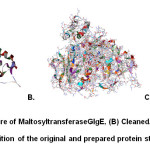 |
Fig1: (A) Original structure of MaltosyltransferaseGlgE, (B) Cleaned, prepared and minimized structure, (C) Superimposition of the original and prepared protein structures (RMSD = 0.721 Å) Click here to View Figure |
To create a pharmacophore, the active site of the protein was first identified using the bound maltose. A site sphere whose radius is 8.0 Å and has x, y and z coordinates of 2.855, -31.798, 2.869, respectively, was generated. It was made sure that it encompasses all the amino acids in the active site (Fig. 2). The RMSD between the original and docked poses of maltose (Fig. 3) was 0.7755 Å, which is still within the acceptable range14. Initially, the pharmacophore has a total of 273 features but was further trimmed down to 19 features by clustering the features and keeping cluster centers. It has 6 acceptor (green), 7 donor (violet) and 6 hydrophobe features (light blue).
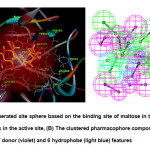 |
Fig2: (A) The generated site sphere based on the binding site of maltose in the protein showing all the amino acids in the active site, (B) The clustered pharmacophore composed of 19 features: 6 acceptor (green), 7 donor (violet) and 6 hydrophobe (light blue) featuresClick here to View Figure |
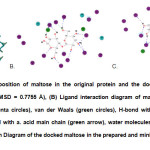 |
Fig3: (A) Superimposition of maltose in the original protein and the docked maltose in the prepared protein (RMSD = 0.7755 Å), (B) Ligand interaction diagram of maltose in the original protein; polar (magenta circles), van der Waals (green circles), H-bond with a. acid side chain (blue arrow), H-bond with a. acid main chain (green arrow), water molecules (aqua blue circles), (C) Ligand Interaction Diagram of the docked maltose in the prepared and minimized protein Click here to View Figure |
Virtual screening allows the rapid selection of a subset of compounds predicted to have significant interactions with the given target out of a large database of molecules. In this study, the use of a pharmacophore was incorporated into virtual screening protocol via pre-screening – a method proven to have the advantage of reducing the size of the database to be docked because docking is the more time-consuming process compared to virtual screening15. Pharmacophore-based virtual screening in DS has been documented to give successful hits in past studies16,17. Virtual screening in DS is done by theScreen Library protocol which has two options for fitting the ligands in the receptor, rigid fitting and flexible fitting, both of which were done in this study with the best hits of the rigid fitting being subjected to the flexible fitting method. The minimum fit value for the compounds to pass the rigid screening test was arbitrarily set to 3.0 while the minimum fit value for the flexible screening was set to 4.0 in order to trim down the number of compounds to be docked to the receptor molecule (Fig.4).
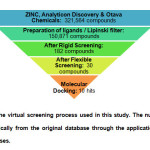 |
Fig4: Summary of the virtual screening process used in this study. The number of compounds was trimmed dramatically from the original database through the application of different filters and screening processes. Click here to View Figure |
The top hits from virtual screening were then subjected to molecular docking using the CDOCKER protocol which is a CHARMm based docking tool that generates random ligand poses and places them in a rigid receptor. In situ ligand minimization was done during the calculation of the binding energy (BE) to remove any ligand van der Waals clashes prior to BE calculations. The docking results showed that the magnitude of binding energy of the top 10 hits were still lower compared tothat of the original bound ligand maltose whose BE is -414.23 kJ/mol (Table 1). Further analysis of the molecular interactions involved in the binding of the compounds with the receptor showed that maltose has the highest number of H-bonds involved (Table 2). However, it is observed that it has a much lower number of van der Waals interaction compared to the other compounds. The top hit ZINC39010596 also has a high number of H-bonds with an additional pi-pi and pi-cation interactions with Tyr357 and Arg392, respectively. Yet, these interactions proved to be insufficient to surpass the high binding energy of maltose.
 |
Table1: Summary of the Properties of Maltose and the Top 10 Hits Click here to View table |
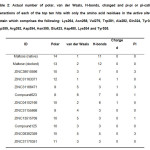 |
Table2: Actual number of polar, van der Waals, H-bonds, charged and pi-pi or pi-cation interactions of each of the top ten hits with only the amino acid residues in the active site of protein which comprises the following: Lys264, Asn268, Val279, Trp281, Ala282, Gln324, Tyr357, Asp359, Arg392, Asp394, Asn395, Glu423, Asp480, Lys534 and Tyr535. Click here to View table |
According to Syson et al.7, the amino acid residues in the active site of the protein include the following: Lys264, Asn268, Val279, Trp281, Ala282, Gln324, Tyr357, Asp359, Arg392, Asp394, Asn395, Glu423, Asp480, Lys534 and Tyr535. Thus, it is essential for the lead compound/s to have significant interactions with these residues. However, only two of the top hits have interactions with all the residues,namely ZINC13515708 and ZINC08382359, which are 7th and 9th in the rank respectively. The top hit ZINC39010596 lacked interaction with Ala282. Maltose, on the other hand, demonstrated interactions with all the residues. It is alsointeresting to note that the following compounds ZINC39010596, ZINC31163371, Compound623, ZINC04102166, ZINC31155668, ZINC13515708 and ZINC08382359 have a common structural feature which is a glucopyranose unit similar to that of maltose. Besides, most of the top hitshave a high number of hydroxyl units, just like maltose, which may be the reason why these compounds have comparable binding energies.The lead compound ZINC39010596 or 5,7-dihydroxy-2-propan-2-yl-8-[(2S,3R,4S,5S,6R)-3,4,5-trihydroxy-6-(hydroxymethyl)tetrahydro-2H-pyran-2-yl]oxychromen-4-one is a glycosylated flavone derivative wherein the phenyl ring was substituted by an isopropyl group (Fig. 5).
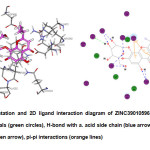 |
Fig5: 3D representation and 2D ligand interaction diagram of ZINC39010596; polar (magenta circles), van der Waals (green circles), H-bond with a. acid side chain (blue arrow), H-bond with a. acid main chain (green arrow), pi-pi interactions (orange lines) Click here to View Figure |
In our desire to search for compounds with higher binding energies than that of maltose, modification of the lead compounds was further employed. The top three lead compounds, ZINC39010596, ZINC31163371 and ZINC31166471 were the ones modified using the De Novo Evolution protocol. In the De Novo Evolution protocol of DS2.5, modifications (i.e. addition of small fragments) are made to an existing ligand scaffold to maximize binding. Ludi scoring was used by the protocol to rank the compounds. Approximately sixty modified molecules were generated by the De Novo Evolution protocol for all three. To trim this down, it was intuitively decided that only the compounds with binding energies higher than -400 kcal/mol were considered. We noticed that some of the modified compounds have high molecular weight, even greater than 500 g/mol, thus violating the Lipinski rule of five. However, recent studies showed that there are actually some good drug candidates that violate this rule18.
We were delighted to find three compounds that have more negative binding energies than the intermediate maltose (BE = -414.23 kcal/mol), all of which originated from ZINC39010596. It is observed that there are similar interactions that occur for the top three modified ligands 1, 2 and 3. All have pi-pi interactions with Trp281, H-bonds with Lys264, Gln324, Arg392, Asp394, Glu423, Asp480, Tyr535 and charged interaction with Tyr535. However, there are still some noticeable differences like for instance; Ligand 1 has an additional H-bond with Lys534 but lacked visible interaction with Ala282. Nonetheless, the similarity in interactions was expected because there are actually only minor differences in the structures of these compounds (Table 3).
 |
Table3: Structures of ZINC39010596 and the top 3 modified ligands Click here to View table |
Furthermore, it is also important to evaluate a priori the pharmacodynamics (e.g. toxicity, degradability, etc.) properties of the molecule. Thus, properties like carcinogenicity and biodegradability were predicted using the Toxicity Prediction (Extensible) protocol of DS2.5. Generally, it is important for a drug to be noncarcinogenic and nonmutagenic. Overall, the calculations showed that the top three ligands are noncarcinogenici and nonmutagenic and are also biodegradable,albeitthey may be irritant to skin and the eyes.
CONCLUSION
Pharmacophore generation, virtual screening, molecular docking (CDOCKER) and de novolead modification were employed in this study for the search of possible inhibitors of MaltosyltransferaseGlgE, a recently validated anti-TB target. Three compounds that have relatively higher binding energies than the intermediate maltose were identified, all of which were derived from ZINC39010596. Toxicity predictions of these compounds also revealed that they are non-carcinogenic, non-mutagenic and biodegradable, athough irritant to the skin and eyes. Experimental bioactivity measurements of potential anti-TB compoundssuch asGlgE inhibitors are underway in our group.
ACKNOWLEDGEMENT
We are grateful to the Emerging Interdisciplinary Research (EIDR) Program of University of the Philippines System (OVPAA-EIDR 12-001-121102) for the use of the Discovery Studio software.
REFERENCES
- Goldmann, D.R.; Horowitz, D.A. American College of Physicians: Complete Home Medical Guide. DK Publishing, Inc. New York, 2003.
- Marwar, A.; Shaker, I. A.; Palawan, H.; Nanadal; Ranjith, M. S.; GokulShankar. J. Basic Clin. Pharm.,2011, 2(1), 27–32.
- (WHO).World Health Organization Global Tuberculosis Report 2012.Geneva, 2012.http://www.who.int/tb/publications/factsheet_global.pdf. (Accessed July 2014).
- Denholm, J. T.; McBryde, E. S. Infect. Drug Resist.,2010,3,63–72.
- Caminero, J. A.,. Lancet Infect. Dis, 2010, 10, 621–629.
- Kalscheuer, R.; Syson, K.; Veeraraghavan, U.; Weinrick, B.; Biermann, K.; Liu, J.; et al.Nat. Chem. Biol.,2010,6(5), 376–384.
- Syson, K., Stevenson, C., Rejzek, M., Fairhurst, S., Nair, A., Bruton, C.; et al.J. Biol. Chem.,2011, 286(44), 38298–38310.
- Marrone, T. J.; Briggs, J. M.; McCammon, J. A. Annu. Rev. Pharmacol. Toxicol.,1997, 37, 71–90.
- Kuntz, I. D.Science,1992, 257, 1078–1082.
- Griffith, R.; Luu, T.;Garner, J.; Keller, P.J. Mol. GraphicsModel., 2005, 23(5), 439–446.
- Discovery Studio v2.5.0.9164 Tutorials. Theory – Prepare Protein. Accelrys Inc., San Diego, CA, 2009.
- Dias, R.; de Azevedo, W. F. Curr. Drug Targets, 2008, 9(12), 1040–1047.
- Lipinski, C. A.; Lombardo, F.; Dominy, B. W.; Feeney, P. J.Adv. Drug Deliv. Rev., 2001, 46,3–26.
- Costakes, G. MMG 445 eJournal, 2005, 1, 1–4.
- Peach, M. L.; Nicklaus, M. C.J. Cheminformatics, 2009, 1(6), 1–13.
- Hou, T.; Xu, X. Curr. Pharm. Des.,2004, 10, 1011–1033.
- Lu, S.H.; Wu, J. W.; Liu, H.L.; Zhao, J.H.; Liu, K.T.; Chuang, C.K.; Lin, H.Y.; Tsai, W. B.; Ho, Y.J. Biomed. Sci., 2011, 18(1), 8.
- Murray, C. W.; Rees, D.C.Nat. Chem.,2009,1, 187–192.

This work is licensed under a Creative Commons Attribution 4.0 International License.
![]()
A New Edition of Web of Science
Journal Impact Factor
2022: 0.5
Five Year: 0.8
Journal is Indexed in

Cabells Whitelist
![]()


