Development of QSAR Model of substituted Benzene Sulphonamide using Multiple Regression Analysis
R.G.Varma and Rashmi Wagde
Department of Chemistry, P.M.B. Gujarati Science College, Indore, India.
DOI : http://dx.doi.org/10.13005/ojc/290447
Article Received on :
Article Accepted on :
Article Published : 25 Dec 2013
Topological indices;Balaban and Balaban related indices
Download this article as:| Copy the following to cite this article: Varma R. G, Wagde R. Development of QSAR Model of substituted Benzene Sulphonamide using Multiple Regression Analysis. Orient J Chem 2013;29(4) |
| Copy the following to cite this URL: Varma R. G, Wagde R. Development of QSAR Model of substituted Benzene Sulphonamide using Multiple Regression Analysis. Orient J Chem 2013;29(4). Available from: http://www.orientjchem.org/?p=1205 |
Introduction:
Carbonic Anhydrase-(II) are important enzymes found in red blood cells, gastric mucosa, pancreatic cells and renal tubes. The catalytic action of CA(II) is fundamental for respiration and transportation of CO2 between metabolizing tissues and organs, pH regulation and homeostasis, CO2 fixation (for algae and green plants), several metabolic biosynthetic pathways, such as glucogenesis, lipid genesis and ureagenesis, bone resorption, calcification and (in vertebrates).
CA-(II) inhibitors have found a wide range of application such as diuretics, antiepileptics, as agents for the treatment of glaucoma and modulators of cancer chemotherapy.
Carbonic Anhydrase is a ubiquitous and physiologically highly relevant isoform. It is highly efficient catalyst for the reversible hydration of carbon dioxide:
CO2 + H2O → HCO3 + H+
At physiological pH, aromatic and heterocyclic unsubstituted sulphonamides
(R-SO2NH2),which are known to inhibit CA-II have an ionisable sulphonamide group
(pKa-6-10). Upon binding the sulphonamide group displaces the water molecule/hydroxide substantially decreases the activity due to steric hindrance(3,4). The aromatic side chains of sulphonamide interacts with hydrophobic amino acid residues in the binding site e.g. Phe 131, Leu 141, Val 143, Ala 145 and stabilize the reaction. Unsubstituted amides such i.e. R-CO2NH2 such as urethane, phenyl carbamate are a second albeit much less potent, class of known CA-II inhibitor. In contrast to sulphonamides the compounds are basic and much weaker. CA-II inhibitor amidst such as SCN–, ClO4–, I– are also weak inhibitor with K ( binding constant) values of 8-13/um(5)
Methodology :
A QSAR , model is developed on the basis of topological indices. The values of topological indices were calculated by applying Graph Theory to the chemical structure. A graph G) G(V,E) is an ordered pair consisting of two sets V) V(G) and E) E(G). Elements of the Set V(G) are called vertices and elements of the set E(G) , involving the binary relation between the vertices, are called edges. By removing all hydrogen atoms from the chemical formula of a chemical compound containing covalent bonds, one obtains the hydrogen-depleted ( or hydrogen-suppressed) molecular graph of that compound. In Graph theory, the atoms are considered as vertices and the bonds as “edges” Vertices are represented by “ and edges by “-”. In this study, the expressions “molecular graph” and “.” “molecule”, “vertex” and “atom”, “edge” and “bond” are used interchangeably. This can be well explained by following example:
 |
Benzene, Molecular graph of Benzene Click here to View figure |
The structural details of parent Benzene sulphonamide and the substituents are given in Table-1.
The methodology used in present investigation is to model the CA-II inhibitory activities of CA-II inhibitors collected from the literature (6-8) . The topological indices are calculated using softwares : Dragon (9) and ACD Labs(10) used for structure optimization.
The molecular descriptors evaluated in present work are: Balabanindex (J)(11) , Balaban-type index : “Z” weighted (Jhetz), mass-weighted (Jhetm), vanderWalls weighted (Jhetv), electronegativity- weighted (Jhete) and polarisability-weighted(Jhetp) Balaban related index (F&G)(12) and the First-order connectivity index or Randic connectivity index(X1)(13)
Balaban index: Balaban Index J (average distance-sum-connectivity index) is defined by(12):
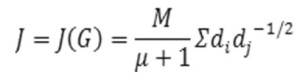
Where M is the number of edges in a graph G,µ is the cyclomatic number of G. di’s (i=1,2,3,4.. is the number of vertices in G) are the distance sums of atoms in G.
Balaban index is extended for the calculation of atom and bond parameters for heteroatom-containing molecules used in computing graph theoretic invariants. This generates a series of Balaban-type index. The different parameters are “Z” weighted (Jhetz), mass-weighted (Jhete), van der Waals weighted (Jhetm), electronegativity-weighted (Jhete) and polarisability-weighted Balaban index. In this approach, the atom and bond weights are computed on the basis of relative atom and bond parameters, using carbon as standard. The new definition of the atom and bond parameters leads to a periodic variation versus the atomic number (Z), with a more natural variation when compared with the parameters defined only by Z. This approach is used to define and compute topological indices based on graph distance. The most used molecular graph descriptors in establishing quantitative structure- property relationships (QSPRS) and quantitative structure-activity relationships (QSAR’s) are topological indices (14-19)
Balaban related index: For those properties where not only the “shape” but also the size of the graph influence the property/activity, two indices related to “J” have been developed. The first of these index is F, defined as:
![]()
This index is able to separate graph size, cyclicity and branching.(20)
The other J-related index “G” is more convenient for correlations. It is defined as:(20)
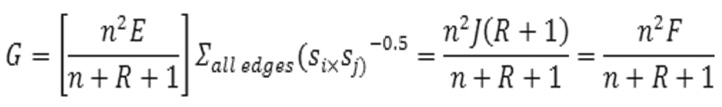
First –order connectivity index ( χ1) : the connectivity-index graph (χ1) of a graph G is defined by Randic as under (21)
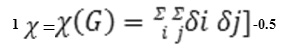
Drug-designing will be performed using correlation analysis employing NCSS (22) software.
The methodology used being the method of maximum-R2 employing multiple regression analysis (23) R2 ( R- squared) is the correlation coefficient between experimental values and model prediction on the test set. Maximum R2 method is used to identify prediction of model. According to Tropsha’s group (24-26) a QSAR model is considered predictive, if the following condition is satisfied :
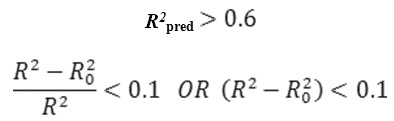
0.85 ≤ k ≤1.15 or 0.85 ≤ k‘ ≤1.15
R2 is the correlation coefficient between experimental values and model prediction on the test set. Mathematical definitions of R20 R2 and K1 are presented clearly in literature (26) and are not described here for brevity.
 |
Table-1 : Basic chemical structures of Carbonic Anhydrase Click here to View table |
Substituent:
(A) Nanofluoro butylsulphonylchloride (C4F9SO2Cl).
(B) Pentafluoro benzene sulphonyl chloride ( C6F5SO2Cl).
Observation:
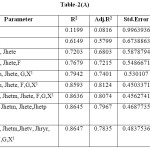 |
Table-2(A): Click here to View table |
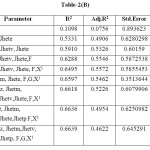 |
Table-2(B): Click here to View table |
Result and Discussion:
Based on the information contained in Table-2(A), we conclude that model 2 to 9 gives R2>0.6 and in Table-2(B) model 4 to 9 shows significant R2.On comparing observations in Table-2(A) and (B), Model 9 in Table-2(A) shows maximum-R2 i.e.0.8647. This model should be considered as best suitable model for inhibition of Carbonic Anhydrase. The Model is as follows:
Estimated Model
-9.48825311691409+.897909585675978*c10+3.07841644442707*c2+
.291542800313685*C3-285691416656861*C4+3.99506176564937E-02*C5+
2.25540907627915*C6+.317756734972618*C7+.422514045832399*C8-
5.33886464481961E-02*C9
All these observations indicates that substituent nanofluoro buty1 sulphony1 chloride(C4F9SO2Cl).
Acknowledgement:We express our sincere gratitude towards Late Dr. P.V.Khadikar.
References:
- Varma, R.G., Wagde, R. J. of Ultrascientist of Physical Sc., 25(1), April (2013).
- Supuran, C.T., Scozzafava, A.Exp. Opin.Theripeutent., 12, 217, (2002)
- Thakur,A.,Thakur,M., Khadikar,PV., Supuran,C.T.,Bioorg.Med.Chem.,12, 789-793 (2004)
- Agrawal,V.K.,Khadikar, P.V., Bioorg.Med Chem.Lett., 13, 447-453 (2003)
- Verpoorte,J.A.,Mehta,S.I.,Edsau, J.T.Biol.Chem., 244, 1421-1429 (1967).
- Supuran, C.T., Scozzafava, A., Menabuoni L., Mincionl.F., Brigantti., Minicionl.C.T., Eu.J.Pharm.Sci. 8, 317-328 (1999)
- Gao,H.,Bajorath, J.Mol.Diversity, 4, 115-130 (1999).
- Clare, B.W., Supuran, C.T., Bioors.Med.Chem.13, 2197-2211 (2005).
- DRAGON
- ACD Labs.
- Balaban, A.T., Basak,S. Beteringhe,A., Mills,D.; Supuran.C.T.,Mol. Discovery, 8, 401-412 (2004)
- Ovidiu Ivanciuc, Teodora Ivanciuc, and Alexandru T. Balaban, J.Chem.Inf.Compit.Sci., 38, 395-401 (1998).
- Mattioni,B.E.,Jurs,P.C.J.Chem.inf.Comput.Sci., 42, 94-102 (2005).
- (a) Balaban, A.T. Applications of Graph Theory in Chemisty.
- J. Chem. Inf. Comput, Sci. 25, 334-343 (1985) .(b) Balaban, A.T. Using Real Numbers as Vertex Invariants for third Generation Topological Indexes. J. Chem. Inf. Comput. Sci. 32, 23-28 (1992).
- (c) Balaban, A. T. Lowering the Intra-and Intermolecular Degeneracy of Topological Invariants. Croat. Chem. Acta 66, 447-458 (1993) (d) Balaban, A.T. Local versus Global ( i.e. Atomic versus Molecular) Numerical Modeling of Molecular Graphs. J. Chem. Inf. Comput. Sci,. 34, 398-402 (1994) (e) Balaban, A.T. Real Number Local (Atomic) Invariants and Global ( Molecular) Topological Indices. Re V. Roum. Chjim., 39, 245-257 (1994). (f) Balaban, A. T. Local (Atomic) and Global ( Molecular) Graph-Theoretical Descriptors. SAR QSAR En Viron. Res., 3, 81-95 (1995).
- Trinajstic, N. Chemical Graph Theory, 2nd ed.; CRC: Boca Raton, FL. (1992)
- Hosoya, H. A Newly Proposed Quantity Characterizing the Topological Nature of Structural Isomers of Saturated Hydrocarbons. Bull. Chem.. Soc.Jpn. 44, 2332-2339 ( 1971)
- Randic, M. On Characterizatoin of Molecular Branching. J. Am. Chem.. Soc. 97, 6609-6615 (1971).
- (a) Kier, L.B. , Hall , L. H. Molecular Connectivity in chemistry and Drug Research; Academic: New York ( 1976). (b) Kier, L.B., Hall, L.H. Molecular Connectivity in Structure-Activity Analysis; Research Studies: Letchworth (1986).
- Schultz, H.P. Topological Organic chemistry. 1. Graph Theory and Topological Indices of Alkanes. J. Chem. Inf. Comput. Sci. 29, 227-228 (1989).
- Balaban, A.T., Khadikar, P.V. , Supuran, C.T., thakur, A, thakur, M., Bioorg. Med. Chem. Lett. 15, 3966-3973 (2005)
- Randic, M, J. Am. Chem.Soc. 97, 6609-6615 (1975).
- NCSS.
- Singh, J., Shaik, B., Singh., S., Agrawal, V.K. , Khadikar, P.V., Deeb O., Supuran, C.T., Chemical biology and drug design (2005).
- Shen, M., Beguin, C., Golbraikh, A., Stables. J. Stables, J. Kohn, H. and Tropsha, A. J. Med. Chem. 47, 2356-2364 (2004).
- Tropsha, A. Gramatica, P. Gombar, V.K., QSAR & Comb. Science. 22, 69-77 (2003).
- Golbraikh, A. and Tropsha, A. J. Mol. Graph Mod., 20, 269-276 (2002).

This work is licensed under a Creative Commons Attribution 4.0 International License.
![]()
A New Edition of Web of Science
Journal Impact Factor
2022: 0.5
Five Year: 0.8
Journal is Indexed in

Cabells Whitelist
![]()


