Crystallographic Evidence for the Host-Guest Interaction of Metallamacromolecules
Cheng-Hua Lee, Chia-Yuan Huang and Pounraj Thanasekaran*
Institute of Chemistry, Academia Sinica, Taipei 115, Taiwa
DOI : http://dx.doi.org/10.13005/ojc/290401
Article Received on :
Article Accepted on :
Article Published : 01 Dec 2013
Metallamacromolecules offer opportunities to create structural diversity and interesting properties based on their unique frameworks and host-guest chemistry. Various types of assemblies can be created by the appropriate choice of the predesigned organic ligands containing various backbones and connectivity information and metal centers. These macrocyclic hosts contain a large and tunable hydrophobic inner cavity, which can be able selectively to recognize guest molecules. The goal of this highlight review is to describe the synthetic routes for the preparation of metallamacromolecules including stepwise and self-assembly strategies as well as their molecular recognition properties.
KEYWORDS:Metallamacromolecules;macrocyclic,hostguest chemistry
Download this article as:| Copy the following to cite this article: Lee C. H, Huang C. Y, Thanasekaran P. Crystallographic Evidence for the HostGuest Interaction of Metallamacromolecules. Orient J Chem 2013;29(4) |
| Copy the following to cite this URL: Lee C. H, Huang C. Y, Thanasekaran P. Crystallographic Evidence for the HostGuest Interaction of Metallamacromolecules. Orient J Chem 2013;29(4). Available from: http://www.orientjchem.org/?p=991 |
Introduction
Macrocyclic host molecules such as calixarenes, cucurbiturils, and cyclodextrins act as catalytical nanoreactors,1 drug delivery agents,2 or molecular detectors.3 The successful application of these molecules often requires functionalization or derivatization with active functional groups. However, their preparation remains very challenging and often requires multistep syntheses that lead to overall low yields. Metallamacromolecules are a class of unique supramolecules, which have space inside to encapsulate cationic, neutral, or anionic guests.4,5 Generally, they do not expose them to the outside environment but bind with them via covalent or non-covalent bond interactions.6 The ability of these metal-organic hosts with the capability of binding guests strongly and selectively is difficult and still remains a challenge for chemists.7 The size and shape of the host structures could be fine-tuned by adjusting or modifying the ligand structure and the preferred geometry of the metal ions. The compounds of palladium(II) and platinum(II) with multidentate pyridine-based ligands were of the first reported cyclic hosts because their square planar geometry provides corners for the formation of different sizes and shapes.8 The simplicity of self-assembly has resulted in a plethora of self-assembled complexes having structures containing molecular triangles, squares, higher-order cyclic species, and closed structures with nanometer-sized cavities. Many impressive examples of metallasupramolecular hosts can be found in studies reported by Lehn,9 Fujita,10 Sauvage,11 Stang,12 Mirkin,13 Hupp,14 Lu,15 Jin16 and other groups.17 Numerous types of metallacyclic hosts have been constructed in a step-wise or one-step synthetic route by using metallo-corner building blockswith bidentate rigid or semirigid linkers and difunctional pyridyl linkers. The preparation of metallacyclic hosts and the investigation of their complexations have produced many insights into significant noncovalent binding mechanisms. The formation and stability of complexation depend on the magnitude and directions of intermolecular interactions in the host−guest assembly. Several examples of systems that exploit aromatic interactions (e.g., π···π, CH···π, anion···π, etc.) and hydrogen bonding interactions have been shown in the creation of host−guest assemblies. Single-crystal X-ray analysis gives information about topology and mode of interaction of the host-guest assemblies in the solid state, which is crucial for thermal stability and kinetics of formation and decomposition of such materials.18 In addition, computational studies are helpful for steering host-guest assembly into prescribed crystal architectures based on well-defined structure directing non-covalent bonding interaction.19 Therefore, the quantitative knowledge of host-guest assembly is of considerable importance for understanding the nature of relevant noncovalent interactions, including those occurring in biological systems. Herein, for the sake of brevity, we highlight some specific examples of synthetic approaches and host behavior of discrete metallacyclic compounds in the solid state.
Preparation and Host-Guest Studies
The use of shape-specific designed ligands such as 1,3-bis(benzimidazol-1-ylmethyl)-4,6-dimethylbenzene L1, 1,3-bis(benzimidazol-1-ylmethyl)-2,4,6-trimethylbenzene L2, or 1,4-bis(benzimidazol-1-ylmethyl)-2,3,5,6-tetramethylbenzene L3 with different metal salts to form a series of metallacyclic structures [Ag2L12](BF4)2 (1), [Ag2L22](CF3SO3)2 (2) and [CF3SO3−ÌAg2L32]CF3SO3(3), [CF3SO3– ÌAg2L33]CF3SO3 (4), [ClO4–ÌCu2L24](ClO4)3 (5) and [4H2OÌNi2L24Cl4]·6H2O (6), respectively (Scheme 1).20
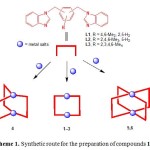 |
Scheme 1. Synthetic route for the preparation of compounds 1–6. Click here to View figure |
X-ray diffraction analysis reveals that in proceeding from 1 to 6 the molecules display an increasingly regular shape, especially with respect to the inner cavity. The cavity in 1–3, however, is arguably not a rectangular box since all the sides are not truly face to-face parallel. The CH3CN solvent molecules and BF4− anions in 1 are found to locate around the metallacycle with almost negligible Ag···F (3.401 Å) and Ag···N (3.021 Å) interactions. In 2, one CF3SO3− anion is dangling on both sides of the molecule by weak Ag···O interaction. On the other hand, one CF3SO3− anion is unambiguously located inside the rectangular cavity of 3 to generate a [CF3SO3−ÌAg2L32]+ cation, although it is crystallographically disordered (Fig. 1). In contrast, the structure of 4 is a prismatic box in which two Ag+ ions are linked by three L3 ligands in a trigonal fashion while the structures of 5 and 6 are tetragonal prismatic cages constructed by two square planar Cu2+ or Ni2+ ions linked by four L2 ligands. Compound 4 hosts one triflate anion, which are weakly interacting with two Ag+ ions (2.543 Å). Two disordered ClO4– anions are alternately arranged inside and outside of 5, thereby linking the molecules into a one-dimensional column with axial Cu···O interactions (2.388 and 2.659 Å), producing polycages. In neutral 6, four symmetrically arranged water molecules are accommodated inside the cavity, which are well ordered and held in place by weak O-H···Cl (O···Cl, 4.555 Å) hydrogen bonds (Fig. 2). Thus, the diversity of the guest molecules endows the structures with tunable inclusion properties and also makes it ambiguous.
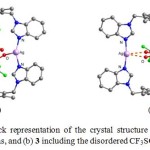 |
Figure 1. Ball and stick representation of the crystal structure of (a) 2 showing two dangling CF3SO3– anions, and (b) 3 including the disordered CF3SO3− guest. Click here to View table |
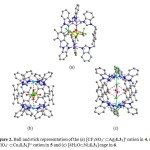 |
Figure 2. Ball and stick representation of the (a) [CF3SO3– ÌAg2L33]+ cation in 4, (b) [ClO4– ÌCu2L34]3+ cation in 5 and (c) [4H2OÌNi2L34] cage in 6. Click here to View figure |
Building on well-established methods for assembling metallosystems incorporating b-diketone ligands,21 Lindoy’s group constructed a large discrete triangular subcomponent. Treatment of β-diketonate ligand (L4H2 = 1,1¢-(4,4¢-biphenylene)bis-3,3-dimethylpentane-1,3-dione) in warm pyridine solution with cobalt(II) acetate tetrahydrate in pyridine afforded the neutral trinuclear CoIII compound, [Co3L43(py)6]·5.55py·0.6H2O (7, py = pyridine) (Scheme 2).22
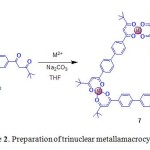 |
Scheme 2. Preparation of trinuclear metallamacrocycle 7. Click here to View figure |
Single-crystal structural analysis revealed that the β-diketonate ligands in 7 were not only bridged by three Co(III) ions but also formed a neutral equilateral triangle (Fig. 50). In addition, the pyridine coligands were axially coordinated to pseudooctahedral mode of CoIII ions. In the solid state structure of 7, a triangular void space was found about 118 Å2 in which the pyridine guest molecule was disordered in this void (Fig. 3). Compound 7 was claimed as the largest neutral M3L’3 triangle so far characterized structurally.23
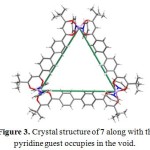 |
Figure 3. Crystal structure of 7 along with the pyridine guest occupies in the void. Click here to View figure |
The characteristics associated with the recognition of molecular rectangles with respect to the planar aromatic molecules and the Ag ion was reported. The alkoxy- or thiolato-bridged molecular rectangles [{(CO)3Re(m-ER)2Re(CO)3}2(m-L5)2] (8–11) (8, -OC8H17; 9, -OC12H25; 10, ER = -SC4H9; 11, ER = -SC8H17) were prepared by the reaction of Re2(CO)10 with the 4,4′-bipyridine (L5, bpy) in the presence of higher aliphatic alcohols or a mercaptan under solvothermal reaction (Scheme 3).24 The more hydrophobic nature of rectangles containing a dodecyl group enhanced their solubility in less polar solvents compared to those carrying an octyl and a butyl groups.
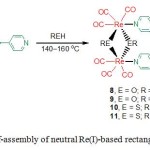 |
Scheme 3. Self-assembly of neutral Re(I)-based rectangles 8-11. Click here to View figure |
Using X-ray crystallographic studies, the rectangular architecture of 10 was confirmed by the distances between Re1···Re2 (3.787 Å) and Re1···Re2A (11.560 Å). The two bpy ligands in 10 are arranged in a face-to-face mode with a weak p-p stacking interaction (both centroid···centroid distances are 3.730 Å), which significantly stabilize the structure of 10. Single crystals of [10·pyrene] suitable for X-ray crystallographic analysis were grown up by the slow evaporation of solvent from an acetone solution of 10 in the presence of pyrene at 25 °C. It was found that the bpy ligands in 10 interact with pyrene via CH···p interactions. The face of the pyrene guest sits over the edges of the bpy linkers, nearly orthogonal with H(pyridyl)···C(pyrene) distances of 2.769-3.295 Å and a dihedral angle of 95° (Fig. 4). Crystal packing studies showed that the host-guest pairs, 10·pyrene, are arranged in a stair-like fashion, in which the guest pyrene molecules are not located within the molecular cavity of 10. This is an example of a CH···p interaction that is rarely designed into a host-guest pair.
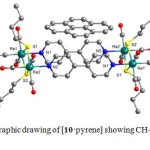 |
Figure 4. Crystallographic drawing of [10·pyrene] showing CH-p interactions in the solid state. Click here to View figure |
Single crystals of [{10·(Ag+)2(NO3–)2(C3H6O)2}(C3H6O)] suitable for X-ray crystallographic study were obtained by dissolving the host 10 with the guest AgNO3 in acetone, followed by slow evaporation at room temperature. The thiolato groups of 10 were firstly coordinated to a Ag atom [Ag1-S1, 2.4405 Å], which was further linked by two NO3– ions with Ag-O distances of 2.308-2.542 Å in addition to the coordination of one acetone molecule [Ag1-O10, 2.432 Å]. The intrinsic affinity of the sulphur atom towards the Ag(I) ion together with the Ag-ONO2 interactions preceded to the formation of a one-dimensional supramolecular array via S···Ag···O connections. Furthermore, two adjacent Ag salts were connected together through NO3– ions with Ag···Ag distances of 4.05 Å, indicating weak argentophilic interactions (Fig. 5).
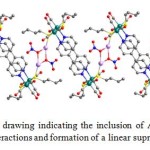 |
Figure 5. Crystallographic drawing indicating the inclusion of AgNO3 moieties by host 10 through Ag-S side-arm interactions and formation of a linear supramolecular array. Click here to View figure |
The highly specific shape, the structural design or modification of the framework for the inclusion phenomena is difficult. Fujita and co-workers have utilized an ethylenediamine (en) derivative or pyridyl “capping” ligands to enforce the 90° cis geometry around square-planar coordinated Pd(II) or Pt(II) ions. The reaction of six (en)Pd2+ and four exo-tridentate ligands such as 1,3,5-tris(4-pyridyl)triazine (L6) or its derivative led to the formation of M6L4-type coordination cages (12–14) (Scheme 4).25 The judicious choice of nitrate counter ions endows compounds 12–14 with high solubility in water.
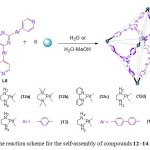 |
Scheme 4. The reaction scheme for the self-assembly of compounds 12-14. Click here to View figure |
X-ray crystallographic study revealed that the four triazine panels in 12–14 occupy alternate faces of the octahedron and generate a very large hydrophobic cavity with a diameter of approximately 2 nm. The cationic cage (overall charge: 12+) is remarkably stable, encapsulates a variety of organic molecules. The unique and efficient binding of large guests by cage 12a has been well reported previously.26,27 Unlike the 12a·(o-carborane)4 complex, which has been never crystallized, 12c·(o-carborane)4, 12c·(1-adamantanol)4, 12c·(diphenylmethane)2, 12c·(tri-tert-butylbenzene), 12c·(1,2-bis(4-methoxypheny)-1,2-ethanedione)2, and 12c·(tetrabenzylsilane) were easily crystallized by allowing its aqueous solution or organic solvents to stand at room temperature for a few days.26d The crystal structure of 12c·(o-carborane)4 revealed that four guest molecules generate (o-carborane)4 aggregates in the cavity of 12c (Fig. 6a). In the solid-state structure of 12c·(o-carborane)4, it was difficult to distinguish carbon from boron atoms since the guest molecules were believed to spin at low temperature. However, 1H NMR study elucidates that guest molecules are oriented in such a way that positive CH is outward while negative (BH)n part is pointed inward.
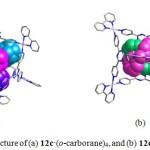 |
Figure 6. Crystal structure of (a) 12c·(o-carborane)4,and (b) 12c·(diphenylmethane)2. Click here to View figure |
The guest molecules in 12c·(1-adamantanol)4 are located at a fixed geometry without disordered. The hydrophobic adamantyl groups are pointed inside, whereas hydrophilic hydroxyl groups are pointed outside. In 12c·(diphenylmethane)2, two diphenylmethane guest molecules are located orthogonally inside the cavity of 12c through CH-p and p-p interactions (3.3-3.6 Å) between two aromatic rings of guests (Fig. 6b). The guest molecule 12c·(tri-tert-butylbenzene) was disordered and triply located with 0.33 occupancy for each. The benzene ring of the guest was located on the triazine ring of one ligand in 12c with a distance of 4.5 Å. In the crystal structure of 12c·(tetrabenzylsilane), each portal accommodated one phenyl group of the guests. In addition, the guest geometries determined by X-ray analyses were consistent with the spectroscopic observation of the solution structure. Thus, the use of 2,2′-bipy ancillary ligand in these macrocyles was found to improve the crystallity of host-guest complexes.
The key to success with the covalently bonded dimetal units is that some coordination sites could be blocked by using non-labile bridging ligands such as N,N‘-di(p-anisyl)formamidinate (L7, DAniF), giving a building block [M2(DAniF)n](4–n)+. With such a building block and linkers, a variety of discrete dimetal-containing macrocycles have been synthesized.28 For example, when cis-Mo2(DAniF)22+ as a vertex building block and was treated with p– or m-trifluoromethyl substituted terephthaloyldiamidate aromatic unit (L8a and L8b) as the linker, two neutral molecular squares 15a and 15b, respectively, were obtained and were structurally characterized.29 X-ray studies of 15a showed that the central square area, approximately 10 ´ 10 Å2, is fenced by eight p-trifluoromethylphenyl groups that create an open cavity. The two p-CF3C6H4 groups on each edge of the square 15a were on the same side of the molecular plane but they alternated from edge to edge by being either above or below the plane. However, the meta position of the CF3 moieties in 15b introduced an additional variable geometry, although it displayed the orientations as that of 15a.
Macromolecule 15b was considered as a class of bowl-shaped molecule in which four aryl groups on the bottom of the square were oriented inward, whereas the four on the top of each molecule were oriented outward (Fig. 7). When the two bowls were approached each other, they formed a cavity where two THF molecules could be encapsulated selectively although a mixture of tetrahydrofuran, toluene, and hexane was chosen as guest molecules in addition to intermolecular F….H interactions.
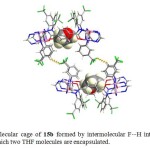 |
Figure 7. Bimolecular cage of 15b formed by intermolecular F…H interactions in the solid state, in which two THF molecules are encapsulated. Click here to View figure |
Self-assembly of giant metallamacrocycles from metal ions and bridging ligands is one of the intriguing topics in current chemistry. The way of the CS2 loss from the metal complexes could be employed in the construction of high nuclearity metallamacrocycles.30 The reaction of ZnCl2 with 3,5-dimethylpyrazolate (L9, K[dmpzdtc]) in a mixture of MeOH and H2O afforded [Zn(L9)2], which was further dissolved in a mixture of DMF and water followed by the elimination of CS2 to give [Zn4(m-dmpz)6(m-OH)2]4 (16). An X-ray structural analysis revealed that compound 16 was consisting of four [Zn4(m-dmpz)6(m-OH)2] units coordinated by sharing pairs of m-dmpz and m-OH2 anions, forming another cyclic structure with a crystallographic four-fold axis running through the centre of the structure (Fig. 8). The distances of the edge and diagonal of 16 were found in the range of 15.7 and 18.0 Å, respectively, indicating a nanosized molecular crown structure. The solvent DMF molecules were located inside and outside of 16 through O2–H···O3 and O1–H···O4 hydrogen bonding interactions, respectively. A very large voids in 16·4DMF with some 1016 Å3 centered at (0, 0, 0.132) and (0.5, 0.5, 0.632) were estimated.
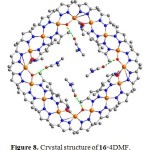 |
Figure 8. Crystal structure of 16·4DMF. Click here to View figure |
Conclusions and Outlook
In this review, the developments of metallamacromolecules from bi-, tri-, tetra- and hexanuclear through octanuclear to hexakaidecanuclear assemblies and their host-guest properties were highlighted. The versatility and efficient synthetic methodologies enable straightforward access to metallamacromolecules, thus allowing the properties to be fine-tuned through a precise chemical alteration of the architectures, opening the way to their use in host-guest chemistry. Studies of synthetic macromolecules with guests have provided significant insight into the nature of relevant non-covalent interactions, including those occurring in biological systems. We demonstrated through some examples that coordination assemblies with well-defined cavities could be used as molecular hosts to encapsulate a variety of guests. According to their solid-state structural studies, the p–p, CH–p interactions, hydrogen bonding and metal–ligand coordination are involved in these systems.
In order to understand the effects on the shape and size of metallamacromolecules with guest molecules, solution-state methods such as NMR spectroscopy and mass spectrometry and solid-state method (X-ray structure analysis) should be integrated for understanding of the host–guest behavior. When the binding interactions are relatively weak and labile, the host–guest complex found in solution and in the crystal may be different. Although the nature of their binding from 1H NMR spectral study in solution is informative, X-ray crystallography provides the clearest structural evidence of the subtle intermolecular interactions. The several mechanisms including guest inclusion, guest exchange, and host–guest interactions are really tedious to follow up because of dynamic and flexible nature of metallahosts. The another challenge to this field is to engineer a system that produces significant changes in selectivity, taking into account the many factors or interactions that may be reinforcing each other or may compete. Thus the functionalization and application of host–guest chemistry of metallamacromolecules still in its fancy and have to be carried out in the coming years. We believe that this highlight review will increase awareness of metallamacromolecules and accelerate the development of host-guest functional materials.
Acknowledgments
The authors thank Academia Sinica and the National Science Council, Taiwan for financial support. We also wish to thank Prof. Kuang-Lieh Lu for his valuable suggestions.
References
- (a) Nimse, S. B. and Kim, T., Chem. Soc. Rev., 42, 366-386 (2013). (b) Joseph, R. and Rao, C. P., Chem. Rev., 111, 4658–4702 (2011). (c) Shenoy, S. R., Pinacho Crisóstomo, F. R., Iwasawa, T. and Rebek, J., Jr., J. Am. Chem. Soc., 130, 5658−5659 (2008). (d) Zelder, F. H. and Rebek, J., Jr., Chem. Commun., 753−754 (2006).
- (a) Ni, X.-L., Xiao, X., Cong, H., Liang, L.-L., Cheng, K., Cheng, X.-J., Ji, N.-N., Zhu, Q.-J., Xue, S.-F. and Tao, Z., Chem. Soc. Rev., DOI: 10.1039/C3CS60261C (2013). (b) Kuykendall, D. W. and Zimmerman, S. C., Nat. Nanotechnol. 2, 201−202 (2007). (c) Memisoglu-Bilensoy, E., Dogan, A. L. and Hincal, A. A., J. Pharm. Pharmacol., 58, 585−589 (2006).
- (a) Chen, G. and Jiang, M., Chem. Soc. Rev., 40, 2254-2266 (2011). (b) Schramm, M. P. and Rebek, J., Jr., Chem. Eur. J., 12, 5924−5933 (2006).
- (a) Dsouza, R. N., Pischel, U. and Nau, W. M., Chem. Rev., 111, 7941–7980 (2011). (b) Martínez-Máñez, R. and Sancenón, F., Chem. Rev., 103, 4419–4476 (2003).
- Clever, G. H., Switchable Host–Guest Interactions of Supramolecular Rings and Cages, in Molecules at Work: Selfassembly, Nanomaterials, Molecular Machinery (ed B. Pignataro), Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany (2012).
- (a) Babine, R. E., Bender, S. L., Chem. Rev., 97, 1359–1472 (1997). (b) Rowan S. J., Cantrill, S. J., Cousins, G. R. L., Sanders, J. K. M. and Stoddart, J. F., Angew. Chem. Int. Ed., 41, 898–952 (2002). (c) Oshovsky, G. V., Reinhoudt, D. N., Verboom, W., J. Org. Chem., 71, 7441–7448 (2006). (d) Schneider, H.-J., Acc. Chem. Res., 46, 1010–1019 (2013).
- (a) Ariga, K., Ito, H, Hill, J. P., Tsukube, H. Chem. Soc. Rev., 41, 5800–5835 (2012). (b) Corbett, P. T., Leclaire, J., Vial, L., West, K. R., Wietor, J.-L., Sanders, J. K. M. and Otto, S., Chem. Rev., 106, 3652-3711 (2006).
- Fujita, M., Yazaki, J. and Ogura, K., J. Am. Chem. Soc., 112, 5645–5646 (1990). (b) Fujita, M., Nagao, S., Iida, M., Ogata, K. and Ogura, K. J. Am. Chem. Soc., 115, 1574–1576 (1993). (c) Fujita, M., Nagao, S. and Ogura, K. J. Am. Chem. Soc., 117, 1649–1650 (1995). (d) Fujita, M., Ibukuro, F., Yamaguchi, K. and Ogura, K. J. Am. Chem. Soc.,117, 4175–4176 (1995).
- Lehn, J. M., Chem. Soc. Rev., 36, 151-160 (2007).
- Fujita, M., Tominaga, M., Hori, A. and Therrien, B., Acc. Chem. Res., 38, 369-378 (2005).
- Forgan, R. S., Sauvage, J. P. and Stoddart, J. F., Chem. Rev., 111, 5434-5464 (2011).
- (a) Cook, T. R., Zheng, Y.-R. and Stang P. J., Chem. Rev., 113, 734-777 (2013). (b) Chakrabarty, R., Mukherjee, P. S. and Stang, P. J., Chem. Rev., 111, 6810–6918 (2011). (c) Leininger, S., Olenyuk, B. and Stang, P. J., Chem. Rev., 100, 853–908 (2000).
- Gianneschi, N. C., Masar, M. S. and Mirkin, C. A., Acc. Chem. Res., 38, 825-837 (2005).
- (a) Slone, R. V., Benkstein, K. D., Bélanger, S., Hupp, J. T., Guzei, I. A. and Rheingold, A. L., Coord. Chem. Rev., 171, 221-243 (1998). (b) Keefe, M. H., Benkstein, K. D. and Hupp, J. T., Coord. Chem. Rev., 205, 201-228 (2000). (c) Lee, S. J. and Hupp, J. T., Coord. Chem. Rev., 250, 1710-1723 (2006).
- (a) Thanasekaran, P., Liao, R. T., Liu, Y. H., Rajendran, T., Rajagopal, S. and Lu, K. L., Coord. Chem. Rev., 249, 1085-1110 (2005). (b) Thanasekaran, P., Lee, C. C. and Lu, K. L., Acc. Chem. Res., 45, 1403-1418 (2012).
- (a) Han, Y. F., Jia, W. G., Yu, W. B. and Jin, G. X., Chem. Soc. Rev., 38, 3419-3434 (2009). (b) Liu, S., Han, Y. F. and Jin, G. X., Chem. Soc. Rev., 36, 1543-1560 (2007). (c) Han, Y. F., Li, H. and Jin, G. X., Chem. Commun., 46, 6879-6890 (2010).
- (a) Amouri, H., Lippert, C. D. B. and Miguel, P. J. S., Chem. Soc. Rev., 40, 4475-4487 (2011). (b) Lindoy, L. F., Park, K.-M. and Lee, S. S., Chem. Soc. Rev., 42, 1713-1727 (2013). (c) Kumar, A., Sun, S. S. and Lees, A. J., Coord. Chem. Rev., 252, 922-939 (2008). (d) Severin, K., Coord. Chem. Rev., 245, 3-10 (2003). (e) Jones, C. J., Chem. Soc. Rev., 27, 289-300 (1998). (f) Swiegers, G. F. and Malefetse, T. J., Chem. Rev., 100, 3483-3538 (2000).
- (a) H. Amouri, C. Desmarets, J. Moussa, Chem. Rev. 2012, 112, 2015–2041. (b) A. S. Mahadevi, G. N. Sastry, Chem. Rev. 2013, 113, 2100−2138.
- (a) Albertí, M., Aguilar, A., Lucas, J. M. and Pirani, F., J. Phys. Chem. A, 114, 11964-11970 (2010). (b) Lamoureux, G. and Orabi, E. A., Mol. Simul., 38, 704-722 (2012). (c) Phung, Q. M., Vancoillie, S. and Pierloot, K., J. Chem. Theory Comput., 8, 883-892 (2012).
- Su, C. Y., Cai, Y. P., Chen, C. L., Smith, M. D., Kaim, W. And zur Loye, H. C. J. Am. Chem. Soc., 125, 8595-8613 (2003).
- Clegg, J. K., Lindoy, L. F., Moubaraki, B., Murray, K. S. and McMurtrie, J. C., Dalton Trans., , 2417–2422 (2004).
- Clegg, J. K., Iremonger, S. S., Hayter, M. J., Southon, P. D., Macquart, R. B., Duriska, M. B., Jensen, P., Turner, P., Jolliffe, K. A., Kepert, C. J., Meehan, G. V. and Lindoy, L. F., Angew. Chem., Int. Ed., 49, 1075-1078 (2010).
- Zangrando, E., Casanova, M. and Alessio, E., Chem. Rev., 108, 4979–5013 (2008).
- Manimaran, B., Lai, L. J., Thanasekaran, P., Wu, J. Y., Liao, R. T., Tseng, T. W., Liu, Y. H., Lee, G. H., Peng, S. M. and Lu, K. L., Inorg. Chem., 45, 8070−8077 (2006).
- (a) Fujita, M., Oguro, D., Miyazawa, M., Oka, H., Yamaguchi, K. and Ogura, K. Nature, 378, 469–471 (1995). (b) Yoshizawa, M., Tamura, M. and Fujita, M. J. Am. Chem. Soc., 126, 6846–6847 (2004).
- (a) Yoshizawa, M., Takeyama, T., Okano, T. and Fujita, M., J. Am. Chem. Soc., 125, 3243-3247 (2003). (b) Kusukawa, T., Yoshizawa, M. and Fujita, M., Angew. Chem., Int. Ed., 40, 1879-1884 (2001). (c) Yoshizawa, M., Takeyama, T., Kusukawa, T. and Fujita, M., Angew. Chem., Int. Ed., 41, 1347-1349 (2002). (d) Kusukawa, T. and Fujita, M., J. Am. Chem. Soc., 124, 13576-13582 (2002). (e) Kawano, M., Kobayashi, Y., Ozeki, T. and Fujita, M., J. Am. Chem. Soc., 128, 6558–6559 (2006). (f) Takaoka, K., Kawano, M., Ozeki, T. and Fujita, M., Chem. Commun., 1625–1627 (2006). (g) Yamauchi, Y., Yoshizawa, M. and Fujita, M. J. Am. Chem. Soc., 130, 5832–5833 (2008).
- Kusukawa, T. and Fujita, M., Angew. Chem., Int. Ed. Engl., 37, 3142-3144 (1998).
- (a) Cotton, F. A., Lin, C. and Murillo, C. A., Acc. Chem. Res., 34, 759-771 (2001). (b) Cotton, F. A., Lin, C. and Murillo, C. A., Proc. Natl. Acad. Sci. U.S.A., 99, 4810-4813 (2002).
- Cotton, F. A., Liu, C. Y., Murillo, C. A. and Wang, X., Inorg. Chem., 45, 2619-2626 (2006).
- Li, H. X., Wu, H. Z., Zhang, W. H., Ren, Z. G., Zhang, Y. and Lang, J. P., Chem. Commun., 5052-5054 (2007).

This work is licensed under a Creative Commons Attribution 4.0 International License.
![]()
A New Edition of Web of Science
Journal Impact Factor
2022: 0.5
Five Year: 0.8
Journal is Indexed in

Cabells Whitelist
![]()


