Theoretical Study of Thermodynamic and Quantum Energy on Carbon Nanotubes Armchair (5,5) and its Derivatives
M. Jamshidi1* , A. Rezaei Bolverdi2, A. Rezaei Bolverdi3, S. Malekian1, Z. Shafiei4 and M. Kahvand1
1Young Researchers Club, Kermanshah Branch, Islamic Azad University, Kermanshah, Iran 2Department of Chemistry, Science and Research Branch, Islamic Azad University,Arak , Iran 3Department of Chemistry, Islamic Azad University, Saveh, Iran 4Department of Chemistry, Islamic Azad University, Shahroud, Iran
Given the increasing use of nanotechnology in basic science and engineering and also discovered the unique properties of nano-tubes and their electronic and magnetic, it will try to take some of the features of this new material examined.In this research, carbon nanotube Armchair (5,5) and 5 derived from its beginning in the next step based on B3LYP and AM1 were optimized. Optimized for greater precision in the results were solely based on B3LYP. The total energy, electron energy and dipole moment was calculated for these nano-tubes.Orbitals on the structure of the nanotubes, they would draw. Derivatives of carbon nanotube Armchair (5,5) with displacement increased to 5 with 5 elements as carbon, phosphorus, was prepared. One result was that by increasing the number of total phosphorus in the composition of the absolute amount of energy will increase.Using graph theory of electron energy levels for the four nano-structure of carbon nanotube derivatives Armchair (5,5) was predictive.
KEYWORDS:nanotube; Armchair; total energy; electron energy
Download this article as:| Copy the following to cite this article: Jamshidi M, Bolverdi A. R, Bolverdi A. R, Malekian S, Shafiei Z, Kahvand M. Theoretical Study of Thermodynamic and Quantum Energy on Carbon Nanotubes Armchair (5,5) and its Derivatives. Orient J Chem 2012;28(3). |
| Copy the following to cite this URL: Jamshidi M, Bolverdi A. R, Bolverdi A. R, Malekian S, Shafiei Z, Kahvand M. Theoretical Study of Thermodynamic and Quantum Energy on Carbon Nanotubes Armchair (5,5) and its Derivatives. Available from: http://www.orientjchem.org/?p=22963 |
Introduction
Carbon nano-tubes, hollow cylinders of graphite sheets that they can be due to their small size (nanometer diameter and micron length) to consider individual molecules that have a transitional period at the end of the axle tubes are.[1]
In addition to the small dimensions of the nanotubes, the high performance and theory can have a thousand times the conventional metallic conductors to carry electric current.[2]
In recent years a wide range of nano-tubes have been examined. Among the important properties of the past research conducted electronic and mechanical properties it can be noted.[3]
Sites in a carbon nanotube can be generally classified into two main groups of single-wall carbon nanotube Sites (SWCNTs) and multi-walled carbon nanotube Sites (MWCNTs) was classified.[4]
Only single-walled carbon nano-tubes and a simple structure (A sheet of regular hexagonal Sites) are formed. Some forecasts suggest that the single layer can be electrically conductive or semi conductive. The electrical conductivity Depending on the exact geometry of carbon atoms is high. Started work on the walls of single of them as a single phenomenon was named to the theory that this will progress step by step.
Interest due to the nanotube Sites and trying to replace single-walled Them in the industry, based on theoretical calculations, laboratory effects on mechanical properties and excellent electrical conductivity such as metals, they are [5].
Quantitative relationships between structure and function, they can manifest with a useful strategic points. Design at the molecular level for membranes with smart features and permeability properties can be controlled using a computer simulation based on the experience of the test molecule design and avoid costly and time-consuming search for the error test[6-7].
One of the best methods of quantum dynamics at the molecular level. The calculation method selected should be done according to what is above is closer to reality, but more time is needed. Basic set of mathematical functions alternative to solve the Schrödinger equation, independent of the timing system is a multi-electron. A method and a suitable base that is extremely useful and accurate B3LYP/6-31G*[8-9].
Material and calculation methods
The nanotube structure using software ChemBioDraw Ultra 12 was plotted and using Gaussian 09W software were optimized based on B3LYP.
After optimization of these nano-structures using GaussView 5.0 software input file was prepared by HyperChem Professional. Ultimate HyperChem Professional software optimization with AM1 method did [10-12].
Table 1: nanotubes studied
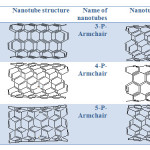 |
Table 1: Click here to View table |
Compounds studied in this section is visible in Table 1 is noteworthy that the compounds have been named as a simple way to ease up the work.
Result and discussion:
Parmatr a separate study in this research as a graph was plotted for nano-structures. In interpreting these results is done.
According to Figure 1 can be seen that with increasing number of phosphorus atoms generally increases the total amount of energy.
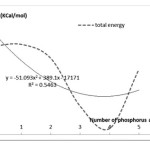 |
Figure 1:Diagram of total energy changes to increase the number of phosphorus atoms in the nanotube Click here to view Figure |
Figure 1: Diagram of total energy changes to increase the number of phosphorus atoms in the nanotube
Another important parameter that was studied is the electron energy. Curved electron energy change is visible in Figure 2.
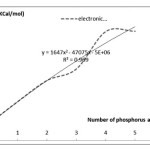 |
Figure 2:Diagram of the electron energy changes increase the number of phosphorus atoms in the nanotube click here to view figure |
As can be observed with increasing electron energy phosphorus increase in nano-tubes. Another important parameter is the dipole moment according to Figures 3 and 4 will be discussed on the curve changes.
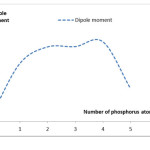 |
Figure 3:Diagram of the dipole moment changes to increase the number of phosphorus atoms in the nanotube Click here to View table |
As can be seen in Figure 3, the dipole moment is close to zero when no phosphorus is combined with the addition of phosphorus can be observed first that the dipole moment increases sharply due to the nanotube is more polar, But after 4 p dipole moment of the greatly reduced and The resultant torque vector due to the neutral phosphorus atoms are polarized. Figure 4 gives us a better view of these changes.
 |
Figure 4:Figure orbitals nano-tubes (a) Armchair (b) 1-P-Armchair (c) 2-P-Armchair Click here to View table |
As we have symmetry orbitals in Armchair nanotubes are relatively high polar moment of this structure is very weak, so the overall outcome is close to zero. But in 1-P- Armchair nanotubes can be observed that due to a phosphorus atom in the compound symmetry severely cluttered and thus greatly enhanced dipole moment[13].
The appropriate regression changes the electron energy is close to an electron energy values using graph theory for nano-tube 4 was expected. The data is given in Table 2.
Table 2: Electron energy values predicted for the new nanotube derivatives
|
Electron energy (Kcal / mol) |
The name of the nanotube |
|
5223158– |
6-P- Armchair |
|
-5248822 |
7-P- Armchair |
|
-5271192 |
8-P- Armchair |
|
-5290268 |
9-P- Armchair |
Conclusion
In this study, the optimal structure Armchair was 6 nanotubes and semi-empirical AM1 method and B3LYP method using the theory of quantum was savable.
The total energy, electron energy and dipole moment of the nanotube was calculated.
With the increasing number of phosphorus atoms in the nano-structure increases the dipole moment is increased until the resultant dipole moment is in one direction.
The increasing number of phosphorus atoms in the nanotubes relative increase in absolute total energy is the electron energy.
Other results of this study was to obtain structural relationship with the electron energy using the QSAR And electron energy levels for the four other nanotubes were predicted on the basis of this method.
Acknowledgments
The authors are grateful for the fiancial support of Young Researchers Club, Islamic Azad University of Kermanshah.
References
- P. L.G.Lauriault, R.Illgen, J.C. Ruiz, M. R. Wertheimer, W.E.S. Unger , Applied Surface Science , 258 , 8448-8454 ,2012
- A.W. Tan, B. Pingguan-Murphy, R. Ahmad and S.A. Akbar , Ceramics International , 38, 4421-4435 , 2012
- C.Cai, J.Chen , Analytical Biochemistry, 332 ,75–83,2004
- K.Imai, T. Akasaka, F.Watari, A.Tanoue, K.Nakamura, K. Suese, H. Takashima, T.Nishikawa, A.Tanaka, S.Takeda , Applied Surface Science , 258 , 8444-8447 ,2012
- D. Gutiérrez-Praena,S. Pichardo ,E. Sánchez ,A. Grilo,A.M. Cameán , “Toxicology in Vitro” ,Vol25 , No8,pp 1883-1888 ,2011
- Z. Yang, K.M.Elrath, J.Bahr, N.A.D’Souza , “Composites: Part B” , Vol43 , No 4 , pp2079-2086 ,2012
- A.Gajewicz, B.Rasulev, T.C. Dinadayalane, P.Urbaszek, T.Puzyn, D.Leszczynska, J.Leszczynski , Advanced Drug Delivery Reviews , xx,xx,2012
- H. Olivier-Bourbigou, L. Magna, D. Morvan , Applied Catalysis A: General , 373 , 1-56 ,2010
- M.T. Baei, M.Moghimi, P.Torabi, A.V.Moradi , Computational and Theoretical Chemistry , 972, 14-19 ,2011
- B.S. Jursic, Journal of Molecular Structure , l 454 , 105-116 , 1998
- S.Peng, Y.Wu, P.Zhu, V.Thavasi, S.G. Mhaisalkar, S.Ramakrishna , Journal of Photochemistry and Photobiology A: Chemistry , 223 , 97-102,2011
- A.Matsuura, H.Sato, W.Sotoyama, A.Takahashi, M.Sakura , Journal of Molecular Structure , 860 , 119-127 ,2008
- M.E. Foster, K.Sohlberg , Computational and Theoretical Chemistry, 984 , 9-12 , 2012

This work is licensed under a Creative Commons Attribution 4.0 International License.
![]()
A New Edition of Web of Science
Journal Impact Factor
2022: 0.5
Five Year: 0.8
Journal is Indexed in

Cabells Whitelist
![]()



{kind=link}