The Influence of Various Solvents on Energetic Property and Stability in C5h4 Divalent Five-Membered Ring: A Dft Studies
Reza Soleymani1*, Khalil Ghesmat-Konandeh2 and Reihaneh Dehghanian Dijvejin2
1Young Researchers Club, Shahre-rey Branch, Islamic Azad University, Tehran, Iran.
2Department of Chemistry, Toyserkan Branch, Islamic Azad University, Toyserkan, Iran.
By using density functional theory (DFT) in B3LYP/6-311++G(d, p) level of theory and conductor-like polarizable continuum modem (CPCM) we examined solvent effects on energy levels in C5H4 for both singlet and triplet states. For this purpose, different solvents, such as gaseous and aqueous solvents, Diethyl ether, Nitro methane, DMSO, Acetonitrile, Methanol, Ethanol, Acetone, Dichloroethane, THF, Aniline, Chlorobenzene, Chloroform, diethyl ether, dichloromethane, toluene, benzene, CCl4, cyclohexane, and heptanes, were used Selectively. The findings suggest that selecting a different solvent changes level of energy, Electrophilicity, Chemical hardness, Chemical potential, and dipole moment for both singlet and triplet states, due to changes in aprotic or perotic properties of solvent. Based on the results, the most stable structure for C5H4 is achieved when the solvent used for both singlet and triplet state is in gaseous phase while the lowest stability is observed when an aqueous solvent is employed for both singlet and triplet states.
KEYWORDS:DFT; CPCM; Singlet–Triplet; Carbenes
Download this article as:| Copy the following to cite this article: Soleymani R, Ghesmat-Konandeh K, Dijvejin R, D. The Influence of Various Solvents on Energetic Property and Stability in C5h4 Divalent Five-Membered Ring: A Dft Studies. Orient J Chem 2012;28(3). |
| Copy the following to cite this URL: Soleymani R, Ghesmat-Konandeh K, Dijvejin R, D. The Influence of Various Solvents on Energetic Property and Stability in C5h4 Divalent Five-Membered Ring: A Dft Studies. Available from: http://www.orientjchem.org/?p=22979 |
Introduction
Carbenes are compounds found in singlet or triplet forms, hence referred to as amphiprotic compounds 1. Such compounds have been the subject of many theoretical and experimental studies and identified as intermediate products in many chemical reactions 2-16. The compounds were first developed and identified in 1991. Two major types of carbenes are Silylene and Germylene. Silylenes are produced through different mechanisms such as thermal decomposition, photochemical mechanisms in silicon hydrates and Organosylanes. They are also key intermediates in CVD 8. Cyclopentadienyldene, a five-membered ring carbene, is a cyclic conjugated compound found in interstellar medium. The stable state of the five-membered ring for the singlet structures Silylenes and Germylenes was first synthesized and identified by Denk et al and their properties were compared to those of other carbenes 9-11. Spin multiplicity of Carbenes and the arrangements of spins are among determinant factors in Carbene reactivity and activity. The multiplicity plays a key role in energy ratio of pi and sigma orbital in base, singlet, and triplet states. In addition, properties of singlet and triplet Carbenes can be examined in terms of electrons and spatial characteristics. Theoretical studies by E. Vessally et al and M.Z. Kassaee et al show that changing substituent plays a key role in reactivity and activity of carbenes in base state 12-16. Research has indicated that type of solvent or substituent (electron donor/acceptor) influence energy level and stability of carbenes. This can be easily verified by examining perturbation orbital diagram. In fact, σ-electron-withdrawing substituents inductively stabilize the nonbonding orbital by increasing its S character. This increases σ-pπ gap, leading to formation of singlet state. In contrast, σ-electron-donating substituents induce a small σ-pπ gap which favors the triplet state.
In cases of substituent change, mesomeric effects may influence stability of carbenes 12-16. Electron-donating substituent increase chemical activity of these compounds and a substituent with lone electron pair adds to this effect. When σ orbital is not stabilized it has almost no charge, leading to increased σ-pπ gap 14-17. Solvent type is also effective in energy level for singlet and triplet C5H4. Different behaviors observed in singlet and triplet states can be attributed to protic or aprotic properties as well as solvent behavior in interaction with C5H4. The present study attempts to explore the effects on solvent type as well as changes in Chemical potential, Chemical hardness, Electrophilicity, and HOMO-LUMO gap.
Electrophilicity was first defined and examined by Parr et al 18. This factor was successfully employed in explaining many systems and describing reactions through organic methods. In some reactions, this factor is regarded as a key player in reaction efficiency for Diels-Alder reactions. Electrophilicity stems from electron structure and is independent of interventions by nuclei. Domingo et al studied relationship between electron effects of substituents, electrophilicity, and Hammett constant for ethylene 18-34. Electrophilicity, largest amount of charge transferred by electrons, chemical hardness, and chemical potential were calculated using Equations (1) to (6), where I denotes ionization potential and A is electron affinity.
μ= (εH +εL)/2 (1)
η=εL -εH (2)
ω=μ2/2η=χ2/2η (3)
χ=-μ=-(бE/бN)V(r) ≈(I+A)/2≈-1/2 (εHOMO+εLUMO) (4)
η=(б2E/бN2)V(r) =(I-A)≈(εLUMO-εHOMO) (5)
∆Nmax=-μ/η (6)
Computational details
Density functional theory (DFT) calculations of C5H4 carbene structure in different solvent were conducted in which geometries, energies and Electrophilicity index values were obtained at the B3LYP/6-311++G(d, p) level of theory. The B3LYP method combines Becke,s three-Parameter exchange (B3) function with the correlation function of Lee-Yong-Parr (LYP) 36. To calculate solvation energies, a popular continuum model of solvation, the conductor-like polarizable continuum model (CPCM) have been used 37. For this purpose “scrf=(cpcm,solvent= solvent name)” key-word used indicates that additional basis functions are to be added to the basis set specified in the route section. Value of energetic parameters are self consistent field calculation used closed-shell (RB3LYP) for the singlet state and unrestricted open-shell (UB3LYP) for the triplet state 38. All calculations were conducted using GAUSSIAN 98W program package 39. However, obtained parameters are calculated in gas phase condition, 298 K temperature and 1 atm pressure.
Result and Discussion:
We first obtained the optimized gaseous structure for C5H4 and identified its energetic parameters (Fig 1). Then, the results were compared to optimum parameters for the same structure in other solvents.
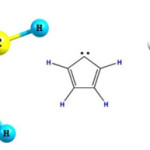 |
Figure 1: Various show of C5H4 carbene with a five membered ring conformation Click here to View figure |
Energetic property
Energy levels in C5H4 were identified in the presence of different solvents. For this purpose, thermal energy, enthalpy, and Gibbs free energy were calculated. Since these parameters in themselves have no use in singlet and triplet states, the gaps for values of these parameters in the two states were obtained. The results for the three parameters followed a nearly similar trend. Tables 1 to 4 and figure 2 present the gap in Gibb free energy for different solvents based on compound stability. Compounds with lower energy levels are expected to be more stable in solvents. As seen here, the lowest energy level, and therefore highest stability for carbene, is observed in solvents like DMSO and water. However, in gaseous solvents or heptanes an increase is observed in Gibbs free energy gaps, resulting in relative instability of structure compared to what observed in aqueous solvents. A similar trend was observed for thermal energy gap and enthalpy gap (see figures 2, 3 and table 4).
Table 1:
Calculated zero point vibration energy (Kcal/mol), dipole moment (Debye), enthalpy (Kcal/mol), gibbs free energy (Kcal/mol), thermal energy (Kcal/mol), enthalpy gaps (Kcal/mol), gibbs free energy gaps (Kcal/mol) and thermal energy gaps (Kcal/mol) for the singlet and triplet states of divalent five-membered ring C5H4 in Water, DMSO, Nitro methane, Acetonitrile, Methanol, Ethanol and Acetone solvents by B3LYP/6-311++G (d, p) level of theory.
|
Spin multiplicity |
Solvent |
ZPVE |
Dipole moment |
G |
H |
E |
∆Gs-t |
∆Hs-t |
∆Es-t |
|
Singlet |
Water |
40.93491 |
1.8085 |
-120969.6288 |
-120949.6568 |
-120950.2491 |
8.342612 |
7.996233 |
7.996232 |
|
DMSO |
40.94798 |
1.7981 |
-120969.5535 |
-120949.5877 |
-120950.1801 |
8.372732 |
8.020705 |
8.020705 |
|
|
Nitro methane |
40.95106 |
1.7888 |
-120969.5222 |
-120949.5589 |
-120950.1512 |
8.382772 |
8.028235 |
8.028235 |
|
|
Acetonitrile |
40.94612 |
1.7840 |
-120969.5253 |
-120949.5545 |
-120950.1468 |
8.374615 |
8.026980 |
8.027608 |
|
|
Methanol |
40.95157 |
1.7862 |
-120969.4989 |
-120949.5325 |
-120950.1249 |
8.385282 |
8.033883 |
8.034510 |
|
|
Ethanol |
40.95895 |
1.7669 |
-120969.4318 |
-120949.4616 |
-120950.0540 |
8.404107 |
8.057100 |
8.057100 |
|
|
Acetone |
40.96422 |
1.7635 |
-120969.3803 |
-120949.4102 |
-120950.0025 |
8.418540 |
8.072160 |
8.072160 |
|
|
Triplet |
Water |
41.22728 |
1.3668 |
-120977.9715 |
-120957.6530 |
-120958.2454 |
|||
|
DMSO |
41.23587 |
1.3613 |
-120977.9263 |
-120957.6084 |
-120958.2008 |
||||
|
Nitro methane |
41.23947 |
1.3582 |
-120977.9049 |
-120957.5871 |
-120958.1795 |
||||
|
Acetonitrile |
41.24039 |
1.3575 |
-120977.8999 |
-120957.5815 |
-120958.1745 |
||||
|
Methanol |
41.24229 |
1.3556 |
-120977.8842 |
-120957.5664 |
-120958.1594 |
||||
|
Ethanol |
41.25142 |
1.3490 |
-120977.8359 |
-120957.5187 |
-120958.1111 |
||||
|
Acetone |
41.25789 |
1.3444 |
-120977.7989 |
-120957.4823 |
-120958.0747 |
Table 2: Calculated zero point vibration energy (Kcal/mol), dipole moment (Debye), enthalpy (Kcal/mol), gibbs free energy (Kcal/mol), thermal energy (Kcal/mol), enthalpy gaps (Kcal/mol), gibbs free energy gaps (Kcal/mol) and thermal energy gaps (Kcal/mol) for the singlet and triplet states of divalent five-membered ring C5H4 in THF, Aniline, Chloro benzene, Chloroform, Diethylether and Gas solvents by B3LYP/6-311++G (d, p) level of theory.
|
Spin multiplicity |
Solvent |
ZPVE |
Dipole moment |
G |
H |
E |
∆Gs-t |
∆Hs-t |
∆Es-t |
|
Singlet |
Dichloro ethane |
41.00018 |
1.7048 |
-120969.0610 |
-120949.0920 |
-120949.6844 |
8.513292 |
8.168167 |
8.168167 |
|
THF |
41.02883 |
1.6611 |
-120968.8250 |
-120948.8624 |
-120949.4553 |
8.586710 |
8.236565 |
8.235937 |
|
|
Aniline |
41.03894 |
1.6480 |
-120968.7384 |
-120948.7770 |
-120949.3694 |
8.613065 |
8.262292 |
8.262920 |
|
|
Chloro benzene |
41.05506 |
1.6088 |
-120968.5408 |
-120948.5737 |
-120949.1661 |
8.662637 |
8.319395 |
8.319395 |
|
|
Chloroform |
41.07633 |
1.5809 |
-120968.3738 |
-120948.4099 |
-120949.0029 |
8.712837 |
8.367712 |
8.367085 |
|
|
Diethyl ether |
41.09534 |
1.5521 |
-120968.2050 |
-120948.2449 |
-120948.8373 |
8.764292 |
8.416030 |
8.416030 |
|
|
Gas |
41.53874 |
1.0374 |
-120964.0560 |
-120944.1147 |
-120944.7070 |
9.913300 |
9.570600 |
9.570700 |
|
|
Triplet |
Dichloro ethane |
41.29637 |
1.3151 |
-120977.5742 |
-120957.2602 |
-120957.8525 |
|||
|
THF |
41.32409 |
1.2943 |
-120977.4117 |
-120957.0989 |
-120957.6913 |
||||
|
Aniline |
41.33421 |
1.2868 |
-120977.3515 |
-120957.0393 |
-120957.6323 |
||||
|
Chloro benzene |
41.35905 |
1.2684 |
-120977.2034 |
-120956.8931 |
-120957.4855 |
||||
|
Chloroform |
41.37848 |
1.2541 |
-120977.0867 |
-120956.7776 |
-120957.3700 |
||||
|
Diethyl ether |
41.39809 |
1.2398 |
-120976.9693 |
-120956.6609 |
-120957.2533 |
||||
|
Gas |
41.86005 |
0.9311 |
-120973.9693 |
-120953.6853 |
-120954.2777 |
Table 3:Calculated zero point vibration energy (Kcal/mol), dipole moment (Debye), enthalpy (Kcal/mol), gibbs free energy (Kcal/mol), thermal energy (Kcal/mol), enthalpy gaps (Kcal/mol), gibbs free energy gaps (Kcal/mol) and thermal energy gaps (Kcal/mol) for the singlet and triplet states of divalent five-membered ring C5H4 in Dichloro methane, Toluene, Benzene, CCl4, Cyclohexane and Heptane solvents by B3LYP/6-311++G (d, p) level of theory.
|
Spin multiplicity |
Solvent |
ZPVE |
Dipole moment |
G |
H |
E |
∆Gs-t |
∆Hs-t |
∆Es-t |
|
Singlet |
Dichloro methane |
41.01257 |
1.6869 |
-120968.9574 |
-120948.9941 |
-120949.5865 |
8.545922 |
8.195777 |
8.195777 |
|
Toluene |
41.21874 |
1.3805 |
-120967.0762 |
-120947.1236 |
-120947.7159 |
9.084317 |
8.736055 |
8.736682 |
|
|
Benzene |
41.23337 |
1.3645 |
-120966.9400 |
-120946.9824 |
-120947.5747 |
9.118830 |
8.776842 |
8.776842 |
|
|
CCl4 |
41.23686 |
1.3613 |
-120966.9155 |
-120946.9604 |
-120947.5528 |
9.127615 |
8.783117 |
8.783117 |
|
|
Cyclohexane |
41.27353 |
1.3231 |
-120966.6470 |
-120946.6981 |
-120947.2905 |
9.211700 |
8.862810 |
8.862810 |
|
|
Heptane |
41.27487 |
1.3023 |
-120966.5177 |
-120946.5607 |
-120947.1537 |
9.234917 |
8.894185 |
8.894185 |
|
|
Triplet |
Dichloro methane |
41.30857 |
1.3058 |
-120977.5033 |
-120957.1899 |
-120957.7823 |
|||
|
Toluene |
41.52931 |
1.1467 |
-120976.1605 |
-120955.8596 |
-120956.4526 |
||||
|
Benzene |
41.54545 |
1.1356 |
-120976.0588 |
-120955.7592 |
-120956.3516 |
||||
|
CCl4 |
41.54791 |
1.1339 |
-120976.0431 |
-120955.7435 |
-120956.3359 |
||||
|
Cyclohexane |
41.57699 |
1.1140 |
-120975.8587 |
-120955.5609 |
-120956.1533 |
||||
|
Heptane |
41.59369 |
1.1028 |
-120975.7526 |
-120955.4549 |
-120956.0479 |
Table 4:Relative enthalpy gaps (Kcal/mol), relative Gibbs free energy gaps (Kcal/mol) and relative thermal energy gaps (Kcal/mol) for the singlet and triplet states of divalent five-membered ring C5H4 in different solvents
|
Singlet-Triplet gaps |
Solvent |
Relative parameters |
||
|
∆Gs-t |
∆Hs-t |
∆Es-t |
||
|
Water |
0.000000 |
0.000000 |
0.000000 |
|
|
DMSO |
0.030122 |
0.024475 |
0.024475 |
|
|
Nitro methane |
0.040162 |
0.032005 |
0.032005 |
|
|
Acetonitrile |
0.032005 |
0.030750 |
0.031378 |
|
|
Methanol |
0.042672 |
0.037653 |
0.038280 |
|
|
Ethanol |
0.061497 |
0.060870 |
0.060870 |
|
|
Acetone |
0.075930 |
0.075930 |
0.075930 |
|
|
Dichloro ethane |
0.170682 |
0.171937 |
0.171937 |
|
|
THF |
0.244100 |
0.240335 |
0.239707 |
|
|
Aniline |
0.270455 |
0.266062 |
0.266690 |
|
|
Chloro benzene |
0.320027 |
0.323165 |
0.323165 |
|
|
Chloroform |
0.370227 |
0.371482 |
0.370855 |
|
|
Diethyl ether |
0.421682 |
0.419800 |
0.419800 |
|
|
Gas |
1.570690 |
1.574370 |
1.574470 |
|
|
Dichloro methane |
0.203312 |
0.199547 |
0.199547 |
|
|
Toluene |
0.741707 |
0.739825 |
0.740452 |
|
|
Benzene |
0.776220 |
0.780612 |
0.780612 |
|
|
CCl4 |
0.785005 |
0.786887 |
0.786887 |
|
|
Cyclohexane |
0.869090 |
0.866580 |
0.866580 |
|
|
Heptane |
0.892307 |
0.897955 |
0.897955 |
|
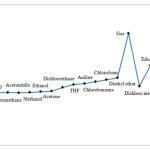 |
Figure 2: Compared obtained results for gibbs free energy gaps, thermal energy gaps and enthalpy energy gaps of five membered ring (C5H4) carbenes structure in various solvents. Click here to View figure |
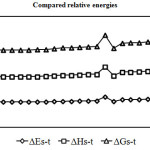 |
Figure 3 :Show comparison Gibbs free energy gaps (Kcal/mol) in various solvents. Click here to View figure |
Dipole moment indices
Tables 1, 2, and 3 present dipole moment values. The results indicate different dipole moments for C5H4 in various solvents. However, in all solvents, dipole moment is greater for singlet state compared to triplet state. To further investigate dipole moments, relative dipole moments were compared for singlet and triplet states. The results found for singlet and triplet states show that replacing aqueous solvent with gaseous ones change dipole moment. For the singlet state, water produces the largest dipole moment while the smallest dipole moment is observed in gaseous solvent. It should be noted, however, that the results are confirmed by dipole moments obtained for the triplet state.
HOMO and LUMO parameters
The HOMO or LUMO levels can be used to determine such parameters as Chemical hardness, Chemical potential, Electrophilicity, and max amount of electronic charge transfer (∆Nmax), which can, in turn, be used to identify or predict chemical properties. We employed quantum mechanics and DFT methods to examine effects of different solvents on singlet and triplet C5H4 structures.
Chemical hardness
Chemical hardness values show that singlet C5H4 in Cyclohexane has the largest chemical hardness while the smallest chemical hardness was observed in acetone. Values found for triplet C5H4 indicate the lowest and highest Chemical hardness in gaseous and aqueous solvents, respectively (Tables 5, 6, 7, 8 and 9). The difference between singlet and triplet states is attributable to differences in interaction between carbene and solvent.
Chemical potential
Chemical potential values show that singlet C5H4 reaches its highest Chemical potential when solved in Ceptane or Cyclohexane while the lowest potential is observed in C5H4 solved in water. For the triplet state, the largest and the smallest chemical potentials were found for aqueous and gaseous solvents, respectively. This indicates totally different chemical potentials for singlet and triplet states (Tables 5, 6, 7, 8 and 9).
Table 5:Calculated εHOMO and εLUMO (eV), chemical hardness (eV), chemical potential (eV), electrophilicity values (eV) and maximum amount of electronic charge transfer in atomic units, for the singlet and triplet states of divalent five-membered ring C5H4 in Water, DMSO, Nitro methane, Acetonitrile, Methanol, Ethanol and Acetone solvents by B3LYP/6-311++G (d, p) level of theory.
|
Spin multiplicity |
Solvent |
εHOMO |
εLUMO |
μ |
η |
ω |
∆Nmax |
|
Singlet
|
Water |
-0.23621 |
-0.11984 |
-0.178025 |
0.116370 |
0.136172 |
1.529819 |
|
|
DMSO |
-0.23620 |
-0.11975 |
-0.177975 |
0.116450 |
0.136003 |
1.528338 |
|
|
Nitro methane |
-0.23616 |
-0.11964 |
-0.177900 |
0.116520 |
0.135806 |
1.526777 |
|
|
Acetonitrile |
-0.23613 |
-0.11974 |
-0.177935 |
0.116390 |
0.136011 |
1.528783 |
|
|
Methanol |
-0.23617 |
-0.11978 |
-0.177975 |
0.116390 |
0.136073 |
1.529126 |
|
|
Ethanol |
-0.23611 |
-0.11961 |
-0.177860 |
0.116500 |
0.135769 |
1.526695 |
|
|
Acetone |
-0.23612 |
-0.11977 |
-0.177945 |
0.116350 |
0.136074 |
1.529394 |
|
Triplet |
Water |
-0.23879 |
0.00239 |
-0.118200 |
0.241180 |
0.028964 |
0.490090 |
|
|
DMSO |
-0.23876 |
0.00234 |
-0.118210 |
0.241100 |
0.028978 |
0.490294 |
|
|
Nitro methane |
-0.23878 |
0.00232 |
-0.118230 |
0.241100 |
0.028988 |
0.490377 |
|
|
Acetonitrile |
-0.23878 |
0.00232 |
-0.118230 |
0.241100 |
0.028988 |
0.490377 |
|
|
Methanol |
-0.23878 |
0.00231 |
-0.118235 |
0.241090 |
0.028992 |
0.490419 |
|
|
Ethanol |
-0.23878 |
0.00226 |
-0.118260 |
0.241040 |
0.029010 |
0.490624 |
|
|
Acetone |
-0.23877 |
0.00222 |
-0.118275 |
0.240990 |
0.029023 |
0.490788 |
Table 6:Calculated εHOMO and εLUMO (eV), chemical hardness (eV), chemical potential (eV), electrophilicity values (eV) and maximum amount of electronic charge transfer in atomic units, for the singlet and triplet states of divalent five-membered ring C5H4 in THF, Aniline, Chloro benzene, Chloroform, Diethylether and Gas solvents by B3LYP/6-311++G (d, p) level of theory.
|
Spin multiplicity |
Solvent |
εHOMO |
εLUMO |
μ |
η |
ω |
∆Nmax |
|
Singlet
|
Dichloro ethane |
-0.23607 |
-0.11964 |
-0.177855 |
0.116430 |
0.135843 |
1.527570 |
|
THF |
-0.23605 |
-0.11955 |
-0.177800 |
0.116500 |
0.135677 |
1.526180 |
|
|
Aniline |
-0.23602 |
-0.11950 |
-0.177760 |
0.116520 |
0.135593 |
1.525575 |
|
|
Chloro benzene |
-0.23594 |
-0.11948 |
-0.177710 |
0.116460 |
0.135586 |
1.525932 |
|
|
Chloroform |
-0.23594 |
-0.11939 |
-0.177665 |
0.116550 |
0.135413 |
1.524367 |
|
|
Diethyl ether |
-0.23590 |
-0.11934 |
-0.177620 |
0.116560 |
0.135333 |
1.523850 |
|
|
Gas |
-0.23592 |
-0.11942 |
-0.177670 |
0.116500 |
0.135479 |
1.525064 |
|
|
Triplet |
Dichloro ethane |
-0.23879 |
0.00199 |
-0.118400 |
0.240780 |
0.029110 |
0.491735 |
|
THF |
-0.23880 |
0.00182 |
-0.118490 |
0.240620 |
0.029174 |
0.492436 |
|
|
Aniline |
-0.23880 |
0.00175 |
-0.118525 |
0.240550 |
0.029200 |
0.492725 |
|
|
Chloro benzene |
-0.23881 |
0.00159 |
-0.118610 |
0.240400 |
0.029260 |
0.493386 |
|
|
Chloroform |
-0.23883 |
0.00146 |
-0.118685 |
0.240290 |
0.029310 |
0.493924 |
|
|
Diethyl ether |
-0.23884 |
0.00132 |
-0.118760 |
0.240160 |
0.029363 |
0.494504 |
|
|
Gas |
-0.23956 |
-0.00296 |
-0.121260 |
0.236600 |
0.031073 |
0.512511 |
Table 7: Calculated εHOMO and εLUMO (eV), chemical hardness (eV), chemical potential (eV), electrophilicity values (eV) and maximum amount of electronic charge transfer in atomic units, for the singlet and triplet states of divalent five-membered ring C5H4 in Dichloro methane, Toluene, Benzene, CCl4, Cyclohexane and Heptane solvents by B3LYP/6-311++G (d, p) level of theory
|
Spin multiplicity |
Solvent |
εHOMO |
εLUMO |
μ |
η |
ω |
∆Nmax |
|
Singlet
|
Dichloro methane |
-0.23608 |
-0.11962 |
-0.177850 |
0.116460 |
0.135800 |
1.527134 |
|
Toluene |
-0.23575 |
-0.11911 |
-0.177430 |
0.116640 |
0.134951 |
1.521176 |
|
|
Benzene |
-0.23579 |
-0.11913 |
-0.177460 |
0.116660 |
0.134973 |
1.521173 |
|
|
CCl4 |
-0.23575 |
-0.11918 |
-0.177465 |
0.116570 |
0.135085 |
1.522390 |
|
|
Cyclohexane |
-0.23573 |
-0.11902 |
-0.177375 |
0.116710 |
0.134786 |
1.519793 |
|
|
Heptane |
-0.23563 |
-0.11912 |
-0.177375 |
0.116510 |
0.135017 |
1.522402 |
|
|
Triplet |
Dichloro methane |
-0.23880 |
0.00192 |
-0.118440 |
0.240720 |
0.029137 |
0.492024 |
|
Toluene |
-0.23896 |
0.00032 |
-0.119320 |
0.239280 |
0.029750 |
0.498663 |
|
|
Benzene |
-0.23898 |
0.00019 |
-0.119395 |
0.239170 |
0.029801 |
0.499206 |
|
|
CCl4 |
-0.23898 |
0.00017 |
-0.119405 |
0.239150 |
0.029808 |
0.499289 |
|
|
Cyclohexane |
-0.23902 |
-0.00008 |
-0.119550 |
0.238940 |
0.029907 |
0.500335 |
|
|
Heptane |
-0.23904 |
-0.00023 |
-0.119635 |
0.238810 |
0.029966 |
0.500963 |
Table 8:Relatives chemical hardness, chemical potential, electrophilicity values and maximum amount of electronic charge transfer in atomic units for the singlet state of divalent five-membered ring C5H4 in different solvents.
|
Spin multiplicity |
Solvent |
μrel |
ηrel |
ωrel |
∆Nmax(rel) |
Dipole moment (rel) |
|
Singlet state |
Water |
0.000000 |
0.000020 |
0.001386 |
0.010026 |
0.7711 |
|
DMSO |
0.000050 |
0.000100 |
0.001217 |
0.008545 |
0.7607 |
|
|
Nitro methane |
0.000125 |
0.000170 |
0.001020 |
0.006984 |
0.7514 |
|
|
Acetonitrile |
0.000090 |
0.000040 |
0.001225 |
0.008990 |
0.7466 |
|
|
Methanol |
0.000050 |
0.000040 |
0.001287 |
0.009333 |
0.7488 |
|
|
Ethanol |
0.000165 |
0.000150 |
0.000983 |
0.006902 |
0.7295 |
|
|
Acetone |
0.000080 |
0.000000 |
0.001288 |
0.009601 |
0.7261 |
|
|
Dichloro ethane |
0.000170 |
0.000080 |
0.001057 |
0.007777 |
0.6674 |
|
|
THF |
0.000225 |
0.000150 |
0.000891 |
0.006387 |
0.6237 |
|
|
Aniline |
0.000265 |
0.000170 |
0.000807 |
0.005782 |
0.6106 |
|
|
Chloro benzene |
0.000315 |
0.000110 |
0.000800 |
0.006139 |
0.5714 |
|
|
Chloroform |
0.000360 |
0.000200 |
0.000627 |
0.004574 |
0.5435 |
|
|
Diethyl ether |
0.000405 |
0.000210 |
0.000547 |
0.004057 |
0.5147 |
|
|
Gas |
0.000355 |
0.000150 |
0.000693 |
0.005271 |
0.0000 |
|
|
Dichloro methane |
0.000175 |
0.000110 |
0.001014 |
0.007341 |
0.6495 |
|
|
Toluene |
0.000595 |
0.000290 |
0.000165 |
0.001383 |
0.3431 |
|
|
Benzene |
0.000565 |
0.000310 |
0.000187 |
0.001380 |
0.3271 |
|
|
CCl4 |
0.000560 |
0.000220 |
0.000299 |
0.002597 |
0.3239 |
|
|
Cyclohexane |
0.000650 |
0.000360 |
0.000000 |
0.000000 |
0.2857 |
|
|
Heptane |
0.000650 |
0.000160 |
0.000231 |
0.002609 |
0.2649 |
Table 9: Relative chemical hardness, chemical potential, electrophilicity values and maximum amount of electronic charge transfer in atomic units for the triplet state of divalent five-membered ring C5H4 in different solvents
|
Spin multiplicity |
Solvent |
μrel |
ηrel |
ωrel |
∆Nmax(rel) |
Dipole moment (rel) |
|
Triplet state |
Water |
0.003060 |
0.004580 |
0.000000 |
0.000000 |
0.4357 |
|
DMSO |
0.003050 |
0.004500 |
0.000014 |
0.000204 |
0.4302 |
|
|
Nitro methane |
0.003030 |
0.004500 |
0.000024 |
0.000287 |
0.4271 |
|
|
Acetonitrile |
0.003030 |
0.004500 |
0.000024 |
0.000287 |
0.4264 |
|
|
Methanol |
0.003025 |
0.004490 |
0.000028 |
0.000329 |
0.4245 |
|
|
Ethanol |
0.003000 |
0.004440 |
0.000046 |
0.000534 |
0.4179 |
|
|
Acetone |
0.002985 |
0.004390 |
0.000059 |
0.000698 |
0.4133 |
|
|
Dichloro ethane |
0.002860 |
0.004180 |
0.000146 |
0.001645 |
0.3840 |
|
|
THF |
0.002770 |
0.004020 |
0.000210 |
0.002346 |
0.3632 |
|
|
Aniline |
0.002735 |
0.003950 |
0.000236 |
0.002635 |
0.3557 |
|
|
Chloro benzene |
0.002650 |
0.003800 |
0.000296 |
0.003296 |
0.3373 |
|
|
Chloroform |
0.002575 |
0.003690 |
0.000346 |
0.003834 |
0.3230 |
|
|
Diethyl ether |
0.002500 |
0.003560 |
0.000399 |
0.004414 |
0.3087 |
|
|
Gas |
0.000000 |
0.000000 |
0.002109 |
0.022421 |
0.0000 |
|
|
Dichloro methane |
0.002820 |
0.004120 |
0.000173 |
0.001934 |
0.3747 |
|
|
Toluene |
0.001940 |
0.002680 |
0.000786 |
0.008573 |
0.2156 |
|
|
Benzene |
0.001865 |
0.002570 |
0.000837 |
0.009116 |
0.2045 |
|
|
CCl4 |
0.001855 |
0.002550 |
0.000844 |
0.009199 |
0.2028 |
|
|
Cyclohexane |
0.001710 |
0.002340 |
0.000943 |
0.010245 |
0.1829 |
|
|
Heptane |
0.001625 |
0.002210 |
0.001002 |
0.010873 |
0.1717 |
Electrophilicity indices
Electrophilicity may be associated with reaction stability. In addition, solvents influence HOMU or LUMO levels in carbenes, and therefore, are expected to change Electrophilicity as well. Our findings, however, indicate that singlet C5H4 reaches the highest level of Electrophilicity when solved in Water, DMSO, or Nitromethane while the lowest level of Electrophilicity is observed in Heptane. When water is used as solvent, greater level of interactions occurs between solvent and carbene and as we shift toward such solvents as DSMO, Electrophilicity is reduced in singlet carbenes. The results for triplet state is, however, completely different. Aqueous solvent results in the lowest level of Electrophilicity while the highest level is achieved through gaseous solvent (see Figures 4 and 5). This can be due to adverse effects of aqueous solvent compared to gaseous solvents
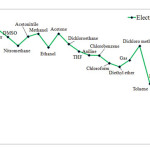 |
Figure 4: Show comparison Electrophilicity in singlet state for various solvents. Click here to View figure |
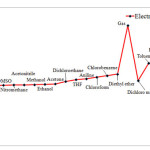 |
Figure 5: Show comparison Electrophilicity in triplet state for various solvents. |
Maximum amount of electronic charge
As mentioned earlier, the largest electron charge received by a system can be calculated using ∆Nmax. The results indicate the largest ∆Nmax for aqueous solvent while the smallest ∆Nmax was found for C5H4 solved in Cylcohexane. The results are totally different for triplet state where the largest ∆Nmax is found for gaseous phase and the smallest ∆Nmax is found when calculations are carried out for aqueous phase. All results confirm various effects of solvent on HOMO-LUMO level and dependent variables.
Conclusion
Quantum mechanics computations using B3LYP/6-311G (d, p) were carried out for singlet and triplet C5H4 through CPCM. The findings can be summarized as follows:
- Examination of parameters revealed that energy levels and dipole moment for singlet and triplet Carbenes vary depending on type of solvent and its protic or aprotic properties.
- Solvent ability in engaging to structure of the compound can influence energy levels as well as HOMO or LUMO levels.
- Aqueous solvent reduces energy level while increasing dipole moment. The opposite effect was observed for gaseous solvent.
- Investigation of HOMO or LUMO levels showed that each solvent has different impacts on this parameter, resulting in different values for Chemical potential, Chemical hardness, Electrophilicity, and ∆Nmax.
- For singlet state, the largest and smallest Electrophilicities were observed for water and Cyclohexane, respectively while in triplet state, the lowest level of Electrophilicity was found for aqueous solvent and the highest level was observed for gaseous solvent.
Acknowledgement
The authors are indebted to Mr. Reza Fazaeli for their interest in this work and many helpful discussions. Moreover, this work was supported by Islamic Azad University Shahre-rey branch.
References
- J. McMurry, Organic Chemistry, 5th Ed., Brooks/Cole, Pacific Grove, CA, (2000) 441.
- A.J. Arduengo, R.L. Harlow, M. Kline, J. Am. Chem. Soc, 113: 361 (1991).
- P.S. Skell, R.C. Woodworth, J. Am. Chem. Soc, 78: 4496 (1956).
- D. Bourissou, O. Guerret, F.P. Gabbai, G. Bertrand, Chem. Rev, 100: 39 (2000).
- W.E. Parham, F.C. Loew, E.E. Schweizer, J. Org. Chem, 24: 1900 (1959).
- W.H. Atwell, D.R. Weyenberg, J. Am. Chem. Soc, 90: 3438 (1968).
- (a) M.D. Sefcik, M.A. Ring, J. Organomet. Chem, 59: 167 (1973); (b) P.P. Gaspar, R. West, in: Z. Rappoport, Y. Apeloig (Eds.), Chemistry of Organic Silicon Compounds, vol. 2, Wiley, Chichester, 1997, p. 2436; (c) R. West, M. Denk, Pure Appl. Chem, 68: 785 (1996); (d) Y. Apeloig, R. Pauncz, M. Karni, R. West, W. Steiner, D. Chapman, Organometallics, 22: 3250 (2003); (e) A. Dhiman, T. Muller, R. West, J.Y. Becker, Organometallics, 23: 5689 (2004).
- C.L. Collins, R.D. Davy, H.F. Schaefer, Chem. Phys. Lett, 171: 259 (1990).
- M. Denk, R. Lennon, R. Hayashi, R. West, A.V. Belyakov, H.P. Verne, A. Haaland, N. Metzler, J. Am. Chem. Soc, 116: 2691 (1994).
- W.A. Herrmann, M. Denk, J. Behm, W. Scherer, F.R. Klingan, H. Bock, B. Solouki, M. Wagner, Angew. Chem, 31: 1485 (1992).
- N. Tokitoh, H. Suzuki, R. Okazaki, K. Ogawa, J. Am. Chem. Soc, 115: 10428 (1993).
- M.Z. Kassaee, S. Arshadi, M. Acedy, E. Vessally, J. Organomet. Chem, 690: 3427 (2005).
- E. Vessally, Heteroat. Chem. 19: 245 (2008).
- E. Vessally, M. Nikoorazm, A. Ramazani, Chin. J. Inorg. Chem, 24: 631 (2008).
- E. Vessally, A. Rezaei, N. Chaliyavi, M. Nikoorazm, Russ. J. Phys. Chem, 81: 1821 (2007).
- E. Vessally, A. Rezaei, N. Chaliyavi, M. Nikoorazm, J. Chin. Chem. Soc, 54: 1583 (2007).
- A. Akbarzadeh, R. Soleymani, M. Taheri, M. Karimi-Cheshme Ali, Oriental Journal of Chemistry, 28: 153 (2012)
- R.G. Parr, L.V. Szentpály, S. Liu, J. Am. Chem. Soc, 121: 1922 (1999).
- E. Chamorro, M. Duque-Noreña, P. Pérez, J. Mol. Struct. (THEOCHEM), 896: 73 (2009).
- L. Meneses, A. Araya, F. Pilaquinga, P. Fuentealba, Chem. Phys. Lett, 460: 27 (2008).
- M. Taheri, R. Soleymani, B. Hosn, H. Rajabzadeh, M.R. Darvish, Oriental Journal of Chemistry, 28: 387 (2012).
- P.R. Campodónico, A. Aizman, R. Contreras, Chem. Phys. Lett, 471: 168 (2009).
- P. Chaquin, Chem. Phys. Lett, 458: 231 (2008).
- C. Makedonas, C.A. Mitsopoulou, Eur. J. Inorg. Chem, 26: 4176 (2007).
- J. Padmanabhan, R. Parthasarathi, V. Subramanian, P.K. Chattaraj, J. Phys.Chem. A, 111: 1358 (2007).
- D.R. Roy, R. Parthasarathi, J. Padmanabhan, U. Sarkar, V. Subramanian,P.K. Chattaraj, J. Phys. Chem. A, 110: 1084 (2006).
- P.R. Campodonico, A. Aizman, R. Contreras, Chem. Phys. Lett, 422: 340 (2006).
- J.L. Moncada, A. Toro-Labbe, Chem. Phys. Lett, 429: 161 (2006).
- H. Aghabozorg , S. Moradi, E. Fereyduni , H. Khani, E. Yaaghubi, Journal of Molecular Structure: THEOCHEM, 915: 58 (2009).
- P.K. Chattaraj, D.R. Roy, Chem. Rev, 107: 46 (2007).
- L.R. Domingo, M.J. Aurell, P. Pérez, R. Contreras, Tetrahedron, 58: 4417 (2002).
- P. Pérez, L.R. Domingo, M.J. Aurell, R. Contreras, Tetrahedron, 59: 3117 (2003).
- R.G. Parr, W. Yang, Density Functional Theory of Atoms and Molecules. Oxford University Press, New York, (1989).
- P. Pérez, A. Toro-Labbé, R. Contreras, J. Am. Chem. Soc, 123: 5527 (2001).
- E. Vessally, E. Fereyduni, M. Kamaee, S. Moradi, J. Serb. Chem. Soc, 76: 879 (2011(.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B, 37: 785 (1988).
- M. Namazian, M. Zakery, M.R. Noorbala, M.L. Coote, Chem. Phys. Lett, 451: 163 (2008).
- W.J. Hehre, L. Radom, P.v.R. Schleyer, J.A. Pople, Ab Initio Molecular Orbital Theory. John Wiley & Sons, New York, (1986).
- M.J. Frisch, et al., Gaussian 98. Gaussian, Inc., Pittsburgh, PA, (1998).

This work is licensed under a Creative Commons Attribution 4.0 International License.
![]()
A New Edition of Web of Science
Journal Impact Factor
2022: 0.5
Five Year: 0.8
Journal is Indexed in

Cabells Whitelist
![]()


