Nmr, Aromaticity and Energetic Property in C6h4o2m (M=C, S, O And N) Dicarbaldehyde Derivatives: A Computational Study
Goldasteh Zarei , Reza Soleymani* and Reyhaneh Dehghaniyan Dejvejen
Department of Chemistry, Tehran Branch, Islamic Azad University, Tehran, Iran
By using density functional theory (DFT) in B3LYP/6-311G(d,p) level of theory, NMR properties, energetic parameters, aromaticity, HOMO and LUMO parameters, were identified for a group of dicarbaldehydes, namely furan-2,5-dicarbaldehyde, pyrrole-2,5-dicarbaldehyde, thiophene-2,5-dicarbaldehyde, and cyclopenta-3,5-diene-1,3-dicarbaldehyde. NMR parameters were examined through GIAO method by calculating NICS at different points for the structures mentioned above. In addition, HOMO and LUMO related properties, including chemical hardness, chemical potential, electrophilicity, and the max amount of electronic charge transferred. Furthermore, to investigate chemical reactivity of the structures, electrostatic potential was evaluated at different parts of each structure.
KEYWORDS:DFT; Aromaticity; NICS; Electrostatic Potential
Download this article as:| Copy the following to cite this article: Zarei G, Soleymani R, Dejvejen R. D. Nmr, Aromaticity and Energetic Property in C6h4o2m (M=C, S, O And N) Dicarbaldehyde Derivatives: A Computational Study. Orient J Chem 2012;28(3). |
| Copy the following to cite this URL: Zarei G, Soleymani R, Dejvejen R. D. Nmr, Aromaticity and Energetic Property in C6h4o2m (M=C, S, O And N) Dicarbaldehyde Derivatives: A Computational Study. Available from: http://www.orientjchem.org/?p=22942 |
Introduction
Thiophene-2,5-dicarbaldehyde, pyrrole-2,5-dicarbaldehyde, furan-2,5-dicarbaldehyde, cyclopenta-3,5-diene-1,3-dicarbaldehyde are compounds from dicarbaldehydes family that previously have been synthesized experimentally 1. Due to their structural characteristics and particular chemical properties, these compounds have found wide applications in different industries. Numerous reports indicate their applications in industry, pharmaceutics, agriculture, chemical industries, as well as uses in variety of mechanisms including complexomety. Given their particular structure, these compounds are capable of going through substitution. Such substitution changes some of their properties, like reactivity or synthetic or thermodynamic parameters 2-10.
The present study attempts to examine the effects of halogen substituent on such parameters as aromaticity scale as well as NMR, HOMO and LUMO related parameters. The first parameter, Nucleus Independent Chemical Shift (NICS), was calculated for different parts of the ring. The difference in this parameter stems from delocalized electron in the ring. Magnetic shielding provides us with exactly those data on delocalized electrons and aromaticity, known as NICS. Conventionally, NICS is defined as the negative value of the absolute magnetic shielding calculated at selected points in vicinity of molecules. Negative values of NICS (magnetic shielding) inside the ring or molecule cages indicate presence of diatropic ring currents or armoaticity while positive values of NICS (lack of magnetic shielding) shows existence of paratropic ring currents or anti-aromaticity 11-13.
NICS is defined as the negative value of the absolute magnetic shielding calculated at the ring centre. In general, NICS values are calculated using virtual nuclei (ghost atoms) at different parts of molecules. The more the negative value of this descriptor the greater is the aromatic character of the molecular system. It is usually better to calculate this parameter in vicinity of the ring center. To calculate this parameters, two virtual points where considered; one at the ring center and another one angstrom above the ring centre (Figures 1 and 2).
 |
Figure 1 Click here to View Figure |
The next parameter, electrophilicity, is a parameter which is well correlated with HOMO and LUMO indices and some reported that it is proportional to compound stability 11. This parameter is calculated using Equation (1). In fact, electrophilicity is proportional to ionization potential and electron affinity. In addition, (2) and (3) can be used to determine chemical potential and chemical hardness. These parameters are correlated with HOMO and LUMO indices as well and therefore differ from one compound to another. Indeed, as electronegativity varies, it is expected that chemical potential changes as well. Increase in electron potential or decrease in chemical hardness can result in a good electrophone species. Equation (4) can be used to calculate the largest amount of charge transferred by electrons 14,15.
Ω=μ2/2η=χ2/2η (1)
χ=-μ=-(бE/бN)VI ≈(I+A)/2≈-1/2 (εHOMO+εLUMO) (2)
η=(б2E/бN2)VI =(I-A)≈(εLUMO-εHOMO) (3)
∆Nmax=-μ/η (4)
Finally, a number of thermodynamic parameters together with electrostatic potentials were evaluated for the structures.
Computational details
All computations were performed in gaseous state, at the pressure of 1 atmospheric and temperature of 298 ̊ K. Pentium IV computer with a 4 Gigabyte of RAM in corei7 processor was used to make computations in XP® operation system. The structures were initially designed through Gauss view 16 and final optimization was carried out using the program package Gaussian 09w 17. B3LYP/6-311G(d,p) level of theory was employed in the computations. Once final optimization was performed, GIAO was used for calculating NICS values and chemical shifts at the different parts of the structures 18. To calculate NICS, two virtual points where considered; one at the ring center and another one angstrom above the ring centre. Chemical shifts for nitrogen nuclei were calculated using TMS as reference. Finally, electrostatic potential was examined for different parts of the structures using Molekel software 19-24.
Results and discussion
Once final optimization was carried out, overall results were extracted. First, thermodynamic parameters were extracted and then other associated parameters were calculated and analyzed.
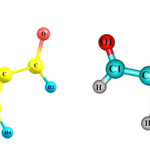 |
Figure 2 Click here to View figure |
Energetic property
Low energy levels indicate higher levels of stability. In other words, reduced level of electronic energy, rotational and vibrational results in more stable structures.
Relative energy and stability
Table 1 summarizes the results obtained through quantum computations. The findings indicate high levels of energy for compounds with Hydrogen substitution and low levels of energy for compounds with Bromine substitution. In addition, when Carbon element is used instead of M atom, a lower level of energy is achieved compared to the case where S, N, or O atoms are used. This can be attributed to the fact that when M is an atom other than Carbon, nonbinding pair of electrons interferes with the resonance space, resulting in higher resonance stability, reduced level of energy, and increased stability (Figure 3).
 |
Figure 3 Click here to View figure |
Dipole moment
Dipole moment is caused by difference in electronegativity of elements, thus, creating difference in magnetic moment. This parameter was examined for the structures studied here. The parameter varies from 2.83 to 6.02 debye. This is attributable to M or halogen substitution. As seen in Table 1, the maximum value for this parameter is achieved when M is substituted with Oxygen while the minimum value is reached when Nitrogen substitutes.
Table 1.
|
Compound |
E0 |
Erel |
Order |
ZPVE |
Dipole moment |
||
|
M |
X |
Entry |
|||||
|
C |
Br |
1a |
-1878927.981 |
225230.932 |
4 |
63.585 |
3.762 |
|
Cl |
2a |
-552442.194 |
1551716.718 |
8 |
63.995 |
3.605 |
|
|
F |
3a |
-326316.710 |
1777842.203 |
13 |
64.970 |
3.835 |
|
|
H |
4a |
-264025.311 |
1840133.601 |
16 |
69.883 |
4.673 |
|
|
N |
Br |
1b |
-1889021.971 |
215136.942 |
3 |
57.456 |
2.987 |
|
Cl |
2b |
-562535.905 |
1541623.007 |
7 |
57.828 |
2.835 |
|
|
F |
3b |
-336408.762 |
1767750.151 |
12 |
58.709 |
3.038 |
|
|
H |
4b |
-274120.996 |
1830037.916 |
15 |
63.765 |
3.964 |
|
|
O |
Br |
1c |
-1901479.093 |
202679.820 |
2 |
49.145 |
5.212 |
|
Cl |
2c |
-574992.900 |
1529166.013 |
6 |
49.525 |
5.002 |
|
|
F |
3c |
-348865.769 |
1755293.144 |
11 |
50.452 |
5.107 |
|
|
H |
4c |
-286579.191 |
1817579.722 |
14 |
55.474 |
6.021 |
|
|
S |
Br |
1d |
-2104158.913 |
0.000 |
1 |
47.132 |
4.134 |
|
Cl |
2d |
-777672.830 |
1326486.082 |
5 |
47.522 |
3.947 |
|
|
F |
3d |
-551546.766 |
1552612.146 |
9 |
48.462 |
4.114 |
|
|
H |
4d |
-489257.966 |
1614900.947 |
10 |
53.463 |
5.118 |
|
HOMO and LUMO indices
HOMO and LUMO indices were studied for the compounds through quantum mechanics calculations. The results are presented in Table 2. These parameters can be used to describe chemical hardness, chemical potential, electrophilicity, and largest amount of transferred charge by electrons for the structures. The findings suggest that dependent parameters vary as HOMO and LUMO values change. Changes in HOMO and LUMO indices totally depend on M and type of halogen substituent attached to it. Figure 4 illustrates HOMO-LUMO gap in the form of DOS spectrum.
Table 2.
|
Compound |
HOMO |
LUMO |
μ |
η |
ω |
∆Nmax |
|
1a |
-0.2684 |
-0.1203 |
-0.1944 |
0.1481 |
0.1275 |
1.3126 |
|
2a |
-0.2705 |
-0.1209 |
-0.1957 |
0.1496 |
0.1280 |
1.3084 |
|
3a |
-0.2698 |
-0.1182 |
-0.1940 |
0.1515 |
0.1242 |
1.2801 |
|
4a |
-0.2652 |
-0.1125 |
-0.1888 |
0.1527 |
0.1168 |
1.2368 |
|
1b |
-0.2619 |
-0.1027 |
-0.1823 |
0.1591 |
0.1044 |
1.1455 |
|
2b |
-0.2646 |
-0.1035 |
-0.1841 |
0.1611 |
0.1051 |
1.1426 |
|
3b |
-0.2643 |
-0.1016 |
-0.1830 |
0.1627 |
0.1029 |
1.1247 |
|
4b |
-0.2598 |
-0.0943 |
-0.1771 |
0.1655 |
0.0947 |
1.0702 |
|
1c |
-0.2778 |
-0.1141 |
-0.1960 |
0.1636 |
0.1173 |
1.1978 |
|
2c |
-0.2808 |
-0.1152 |
-0.1980 |
0.1655 |
0.1184 |
1.1961 |
|
3c |
-0.2813 |
-0.1140 |
-0.1976 |
0.1673 |
0.1167 |
1.1811 |
|
4c |
-0.2767 |
-0.1067 |
-0.1917 |
0.1699 |
0.1081 |
1.1280 |
|
1d |
-0.2762 |
-0.1213 |
-0.1987 |
0.1549 |
0.1275 |
1.2832 |
|
2d |
-0.2799 |
-0.1221 |
-0.2010 |
0.1578 |
0.1280 |
1.2736 |
|
3d |
-0.2807 |
-0.1201 |
-0.2004 |
0.1606 |
0.1251 |
1.2482 |
|
4d |
-0.2787 |
-0.1135 |
-0.1961 |
0.1651 |
0.1164 |
1.1875 |
 |
Figure 4 Click here to View figure |
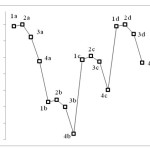 |
Figure 5 Click here to View figure |
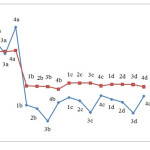 |
Figure 6 Click here to View figure |
Figure 5 depicts the changes in electrophilicity. As seen in the figure, the minimum electrophilicity is reached when M is substituted with Nitrogen.
Aromaticity indices
Aromaticity is directly connected to stability. The more aromatic is a structure the higher levels of stability are expected. Changes in aromatic properties in the compounds were determined using NICS for different parts. The more negative is the NICS value, the higher levels of aromatic stability are expected. The index is, however, only considered for rings.
NICS indices
NICS indices for the structures were calculated using GIAO. The results are presented in Table 3. To investigate NICS values in the compounds we considered two points: one at the ring center and another one angstrom above the ring centre. Figure 6 compares the results. As seen in the results, the parameter varies as M is substituted with another atom or as the substituent X is replaced. The figure indicates that in majority of compounds examined here, NICS value at the ring center is more negative compared to corresponding values obtained at one angstrom above the ring center. Therefore, higher levels of aromaticity are expected at the ring center compared to the space above the center. This can be attributed to higher resonance inside the ring which creates isotropic current.
Table 3.
|
Compound |
Nucleic independent chemical shift (ppm) |
|
|
NICS(0) |
NICS(1) |
|
|
1a |
-3.5548 |
-5.5105 |
|
2a |
-4.0763 |
-5.5643 |
|
3a |
-5.9041 |
-5.6021 |
|
4a |
-2.4991 |
-5.4926 |
|
1b |
-12.4919 |
-10.0374 |
|
2b |
-12.9353 |
-10.0839 |
|
3b |
-14.5509 |
-10.1251 |
|
4b |
-12.1694 |
-10.4342 |
|
1c |
-11.5171 |
-9.6580 |
|
2c |
-11.9401 |
-9.6641 |
|
3c |
-13.4416 |
-9.6572 |
|
4c |
-11.2771 |
-10.0178 |
|
1d |
-11.7664 |
-9.8161 |
|
2d |
-12.1260 |
-9.8148 |
|
3d |
-13.5300 |
-9.8487 |
|
4d |
-11.3356 |
-10.1376 |
1H-NMR indices
Nucleus magnetic resonance (NMR) describes the chemical environment around nuclei and can be used to determine some chemical properties of compounds. In the structures studied here, chemical shift was investigated for hydrogen atom nucleus using DFT-GIAO in B3LYP/6-311G(d,p) level of theory. Isotropic chemical shift values of Hydrogen nuclei were studied as well (Table 4). All results were obtained using TMS as reference.
Table 4.
|
Methods |
Isotropic Chemical Shift (TMS reference) |
|||||
|
B3LYP/6-311G(d,p) |
||||||
|
Entry |
H1 |
H2 |
H3 |
H4 |
H5 |
H6 |
|
1a |
10.40 |
10.22 |
6.99 |
– |
3.53 |
3.53 |
|
2a |
10.46 |
10.20 |
6.91 |
– |
3.50 |
3.50 |
|
3a |
10.48 |
10.23 |
6.96 |
– |
3.45 |
3.45 |
|
4a |
10.32 |
10.32 |
7.28 |
7.28 |
3.54 |
3.54 |
|
1b |
10.10 |
9.90 |
6.68 |
– |
9.20 |
– |
|
2b |
10.14 |
9.87 |
6.58 |
– |
9.06 |
– |
|
3b |
10.16 |
9.90 |
6.38 |
– |
8.82 |
– |
|
4b |
10.02 |
10.02 |
6.80 |
6.80 |
9.31 |
– |
|
1c |
10.23 |
9.99 |
6.88 |
– |
– |
– |
|
2c |
10.26 |
9.96 |
6.80 |
– |
– |
– |
|
3c |
10.26 |
9.96 |
6.72 |
– |
– |
– |
|
4c |
10.10 |
10.10 |
7.00 |
7.00 |
– |
– |
|
1d |
10.31 |
10.13 |
7.25 |
– |
– |
– |
|
2d |
10.36 |
10.10 |
7.16 |
– |
– |
– |
|
3d |
10.43 |
10.14 |
7.10 |
– |
– |
– |
|
4d |
10.25 |
10.25 |
7.47 |
– |
– |
– |
Structural parameters
Once final optimization was carried out, length of structures were determined and analyzed. The results indicate that changing halogen substituent or M atoms leads to changes in bond length at some parts of the structure. Table 5 presents the results. As seen in the table, changes in vicinity of halogen substituent experience more variations as halogen substitution takes place.
Table 5.
|
Entry |
Bond length (Angstrom) |
||||||||
|
C1-C2 |
C5-C6 |
C2-C3 |
C4-C5 |
C2-M |
C5-M |
C3-C4 |
C1-O1 |
C6-O2 |
|
|
1a |
1.466 |
1.463 |
1.357 |
1.362 |
1.503 |
1.500 |
1.451 |
1.213 |
1.211 |
|
2a |
1.466 |
1.462 |
1.357 |
1.362 |
1.500 |
1.502 |
1.450 |
1.211 |
1.214 |
|
3a |
1.466 |
1.458 |
1.358 |
1.359 |
1.502 |
1.504 |
1.445 |
1.211 |
1.214 |
|
4a |
1.463 |
1.463 |
1.361 |
1.361 |
1.501 |
1.501 |
1.449 |
1.213 |
1.213 |
|
1b |
1.460 |
1.459 |
1.397 |
1.400 |
1.362 |
1.365 |
1.402 |
1.213 |
1.214 |
|
2b |
1.460 |
1.458 |
1.396 |
1.400 |
1.362 |
1.365 |
1.401 |
1.213 |
1.215 |
|
3b |
1.460 |
1.454 |
1.399 |
1.397 |
1.361 |
1.368 |
1.396 |
1.213 |
1.215 |
|
4b |
1.457 |
1.457 |
1.398 |
1.398 |
1.364 |
1.364 |
1.453 |
1.214 |
1.214 |
|
1c |
1.467 |
1.466 |
1.373 |
1.379 |
1.357 |
1.359 |
1.415 |
1.206 |
1.207 |
|
2c |
1.468 |
1.465 |
1.373 |
1.380 |
1.354 |
1.357 |
1.414 |
1.206 |
1.208 |
|
3c |
1.468 |
1.461 |
1.375 |
1.377 |
1.354 |
1.360 |
1.408 |
1.206 |
1.208 |
|
4c |
1.465 |
1.465 |
1.376 |
1.376 |
1.356 |
1.356 |
1.415 |
1.207 |
1.207 |
|
1d |
1.468 |
1.468 |
1.377 |
1.382 |
1.735 |
1.739 |
1.411 |
1.209 |
1.210 |
|
2d |
1.468 |
1.467 |
1.377 |
1.383 |
1.735 |
1.738 |
1.410 |
1.208 |
1.210 |
|
3d |
1.468 |
1.463 |
1.379 |
1.380 |
1.737 |
1.738 |
1.405 |
1.208 |
1.211 |
|
4d |
1.465 |
1.465 |
1.381 |
1.381 |
1.737 |
1.737 |
1.410 |
1.209 |
1.209 |
Electrostatic potential map
To evaluate electric potentials in the structures, we first optimized the structures using B3LYP/6-311G(d,p) level of theory and then calculated electric potential for each structure 25. Molecular electronic potential maps (MEPM) determine locations which are more likely to become the target for electrophilic attacks. In fact MEPM determines reactivity for different parts of molecules.
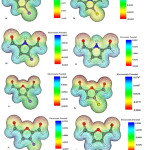 |
Figure 7 Click here to View figure |
As seen in Figure 7, as we approach the blue region electric potential increases while areas near the red region represent smaller electric potentials. Electric potential is negative at the location of Oxygen atoms while for Carbon groups, electric potential becomes positive (i.e. closer to the blue region).
Conclusion
Density functional theory (DFT) and B3LYP/6-311G(d,p) were used to optimize 16 dicarbaldehyde compounds and several parameters, including energetic parameters, aromaticity, HOMO and LUMO indices, and electrophilicity, for these structures were compared to those of the stable and symmetric structure of benzene. The findings can be summarized as follows:
Investigation of energetic level reveals that among the 6a structures studied here, 3-bromothiophene-2,5-dicarbaldehyde has the lowest level of energy, and therefore, is the most stable compounds while cyclopenta-3,5-diene-1,3-dicarbaldehyde is the least stable structure with the highest energy level.
NICS analysis indicates that in all compounds studied here isotropic effect at the ring center is more significant than the effect observed at one angstrom above the ring center. This can be attributed to increased resonance induced by nonbonding pair of electrons is some compounds with M group.
In compounds where M atom is substituted with M, electrophilicity is lower compared to compounds with other substituents.
As halogen substituent or M atoms are changed, bond lengths experience some variations. These variations are larger at regions directly engaged with M atom or halogen substituent. In addition, changing the halogen substituent leads to changes in dipole moment and electrostatic potential both depending on electron donating withdrawing power of substituent.
Acknowledgements
This work was supported by Islamic Azad University Shahre-rey branch and Islamic Azad University Toyserkan branch.
References
- B.L. Feringa, R. Hulst, R. Rikers, L. Brandsma, Synthesis, 15: 316 (1988).
- J. Lewkowski, ARKIVOC, (i): 17 (2001).
- K. B. Kokoh, E. M. Belgsir, Tetrahedron Letters, 43: 229 (2002).
- S.P. Verevkin, V.N. Emel’yanenko, E.N. Stepurko, R.V. Ralys, D.H. Zaitsau, Ind. Eng. Chem. Res, 48: 10087 (2009).
- Sh. Takekuma, K. Tone, M. Sasaki, T. Minematsu, H. Takekuma, Tetrahedron, 63: 2490 (2007).
- H. Adams, N.A. Bailey, D.E. Fenton, P.D. Hempstead, G.P. Westwood, Journal of Inclusion Phenomena and Macrocyclic Chemistry, 11: 63 (1991).
- Sh. Takekuma, S. Hori, T. Minematsu, H. Takekuma, Research Letters in Organic Chemistry, 2009: 5 (2009).
- A. Michaleviciute, G. Buika, J.V. Grazulevicius, F. Tran-Van, C. Chevrot, V. Jankauskas, Molecular Crystals and Liquid Crystals, 468: 107 (2007).
- E. Aqad, P. Leriche, G. Mabon, A. Gorgues, M. Allain, A. Riou, A. Ellern, V. Khodorkovsky, Tetrahedron, 59: 5773 (2003).
- V.A. Knizhnikov, N.E. Borisova, N.Y. Yurashevich, L.A. Popova, A.Y. Chernyadev, Z.P. Zubreichuk, M.D. Reshetova, Russian Journal of Organic Chemistry, C/C of Zhurnal Organicheskoi Khimii, 43: 855 (2007).
- R. Soleymani, H. Rajabzadeh, Asian Journal of Chemistry, 24: 4614 (2012).
- S. Gumus, Comput. Theor. Chem, 963: 263 (2011).
- J. Kruszewski, T.M. Krygowski, Tetrahedron Lett, 13: 3839 (1972).
- R.G. Parr, L.V. Szentpaly, S. Liu, J. Am. Chem. Soc, 121: 1922 (1999).
- E. Chamorro, M. Duque-Norea, P. Perez, J. Mol. Struct. (THEOCHEM), 896: 73 (2009).
- R. Dennington, T. Keith, J. Millam, K. Eppinnett, W.L. Hovell, R. Gilliland, GaussView, Version 3.07, Semichem, Inc., Shawnee Mission, KS, (2003).
- M.J. Frisch et al, GAUSSIAN 09w, Gaussian, Inc, Pittsburgh, PA, (2010).
- Y.J. Beevi, H.T. Varghese, C.Y.Panicker, G. Rajendran, Oriental journal of chemistry, 27: 1109 (2011).
- B. Semire, O.A. Odunola, Oriental journal of chemistry, 25: 841 (2009).
- C.Y. Panicker, H.T. Varghese, A. Chandran, Oriental journal of chemistry, 27: 1771 (2011).
- C.Y. Panicker, H.T. Varghese, B. Harikumar, A. Chandran, Oriental journal of chemistry, 27: 1705 (2011).
- Y.J. Beevi, H.T. Varghese, C.Y.Panicker, G. Rajendran, Oriental journal of chemistry, 27: 1109 (2011)
- M.A. Boughdiri, O. Fliss, T. Boubaker, B. Tangour, Oriental journal of chemistry, 22: 39052 (2006).
- M. Taheri, R. Soleymani, B. Hosn, H. Rajabzadeh, M.R. Darvish, Oriental Journal of Chemistry, 28: 387 (2012).
- P. Politzer, D.G. Truhlar, Chemical Application of Atomic and Molecular Electrostatic Potentials, Plenum, New York, (1981).

This work is licensed under a Creative Commons Attribution 4.0 International License.
![]()
A New Edition of Web of Science
Journal Impact Factor
2022: 0.5
Five Year: 0.8
Journal is Indexed in

Cabells Whitelist
![]()


