Molecular Structure, Experimental and Theoretical Spectroscopic Studies and Quantum Chemical Calculation of Phenoxyacetic Acid and Its P-Chloro Derivative
Anju Srivastav1*, Subha R. Saxena2, Sunil Mishra2 and Y. Singh1
1Department of Chemistry, Udai Pratap Autonomous College, Varanasi - 221 002, India. 2Department of Physics, A. K. A. P. G. College, Varanasi - 221 002, India.
The Fourier Transform Infrared spectra of crystallized phenoxyacetic acid and p-chloro derivative have been recorded in the region 4000-400 cm-1. The geometry was calculated by Hartree-Fock (HF), Density Functional Theory (B3LYP) and Moller Plesset(MP2) Perturbation theory invoking 6-311++g(d,p) basis set. The harmonic vibrational frequency and, infrared intensities have been investigated with the help of B3LYP methods. The scaled theoretical wavenumbers showed very good agreement with the experimental values. The thermodynamic functions of the title compounds were also performed at B3LYP/6-311++g(d,p) level of theory. A detailed interpretation of the infrared spectra of Phenoxyacetic acid and p-chloro derivative of Phenoxy acetic acid have been reported.
KEYWORDS:Vibrational spectra; DFTcalculation; Moller Plesset Perturbation theory
Download this article as:| Copy the following to cite this article: Srivastav A, Saxena S. R, Mishra S, Singh Y. Molecular Structure, Experimental and Theoretical Spectroscopic Studies and Quantum Chemical Calculation of Phenoxyacetic Acid and Its P-Chloro Derivative. Orient J Chem 2012;28(3). |
| Copy the following to cite this URL: Srivastav A, Saxena S. R, Mishra S, Singh Y. Molecular Structure, Experimental and Theoretical Spectroscopic Studies and Quantum Chemical Calculation of Phenoxyacetic Acid and Its P-Chloro Derivative. Available from: http://www.orientjchem.org/?p=23208 |
Introduction
Aryloxyacetic acid are interesting to study by various chemical and physical methods. Phenoxyacetic acid is one of the best known aryloxyacetic acid, which is useful in the treatment of insulin resistance and hyperglycemia which has been investigated by various researchers [1-2]. Phenoxyacetic acid and substituted phenoxyacetic acids have potential biological properties. These acids are widely used in herbicides [3] and pesticide[4] formulations. Recently phenoxyacetic acid invoke much interest among the spectroscopists owing to their novel bioactivity such as anticancer, antitumour; anti-inflammatory [5], antimicrobial, plant growth regulator and inhibillation of tillage etc [6, 7].
Vibrational spectroscopy is a valuable tool for the elucidation of molecular structure [8]. It also provides important information about the intermolecular forces acting between the atoms in a molecule. The vibrational and rotational energies of molecules can be studied by IR and Raman spectroscopy [9, 10 ].The advent of fast computer along with sophisticated computational methods make the task of solving various structural chemical problem a simple. Ab initio DFT computations have recently become an efficient tool in the prediction of molecular structures, harmonic force fields, vibrational frequencies and IR spectral properties of biological compounds [11,12].The calculation of vibrational frequency using DFT provides a promising cost effective approch for calculating vibrational spectra of large molecules. In the recent studies, the harmonic vibrational frequencies for a large number of small and well-studied organic molecules were computed with HF and DFT methods[13-16].
Many researches have been done on the vibrational analysis of phenols [17]. However, for phenoxyacetic acid and p-chloro derivative of phenoxyacetic acid, such a kind of work are limited. Only IR spectral assignments and normal coordinate analysis has been carried out for some phenoxyacetic acid [18-21].The molecular and crystal strucrure of phenoxyacetic acid and p-chloro phenoy acetic acid were reported by Colin H.L.Kennard[22] and by Vijaykumar and Rao [23]. Karthikeyan and Saravanan[24] reported tentative Raman spectral assignments of phenoxyacetic acid and chlorine substituted phenoxy acetic acid with the aid of ab initio calculation HF/3-21G(d) basis set, however some of its assignments was ambiguous. Ab initio HF and DFT calculation of p-chlorophenoxy acetic acid was done by N.Sunaraganeshan and B.Karthikeyan [25] but no MP2 calculation was done. For phenoxy acetic acid neither MP2 nor DFT calculation have been reported. Therefore, the present investigation was undertaken to study the vibrational spectra of phenoxyacetic acid and p-chloro derivative. MP2, ab initio HF and density functional theory calculations have been performed to predict molecular structure of phenoxyacetic acid and p-chloro derivative.
Materials and Methods
Phenoxyacetic acid was prepared by using slightly modified method of Koelsch [26]. The same procedure was followed for substituted phenoxy acetic acid i.e. p-chlorophenoxy acetic acid by using corresponding substituted phenols. Both the compounds i.e. phenoxyacetic acid and p-chloro derivative were recrystallized using water as a solvent.
Computational details
Gaussian 09 package was used for calculating theoretical vibrational frequencies [27]. Density functional theory [28] with the three-parameter hybrid functional (B3) [29] for the exchange part and Lee-yang Parr [LYP] correlation function [30] was used for the computation of molecular structure, vibrational frequency and energies of optimized structure. The atom numbering and the structure which was used for the input was given in figure 1. The bond lengths and bond angles wera taken from the X-ray crystal studies [31-33]. This structure was optimized (Cs) point group using Hartree-Fock, B3LYP and MP2 levels, adopting the standard 6-311++g(d,p) basis set. Raw frequency values computed at B3LYP level contain known systematic error due to the neglect of electron correlation, resulting in overestimation of about 10-12%. Therefore, the obtained frequencies were scaled by an empirical factor 0.98 [27].
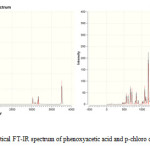 |
Figure 1: Theoretical FT-IR spectrum of phenoxyacetic acid and p-chloro derivative. |
Results and Discussion
Optimized Geometries
The optimized geometry of phenoxyacetic acid and p-chloro derivative are calculated by ab initio HF, DFT and MP2 levels with the 6-311++g(d,p) basis set. The molecular conformation and numbering are illustrated in Fig 2 and bond lengths and angles for phenoxyacetic acid and p-chloro derivative are given in table 1 and 2. Table 1 and 2 compares the calculated bond lengths and angles for phenoxyacetic acid and p-chloro derivative with those of experimentally available from X-ray data for phenoxy acetic acid (23) and p-chloro (24) derivative.
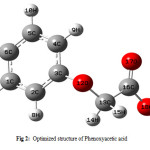 |
Figure 2: Optimized structure of Phenoxyacetic acid. |
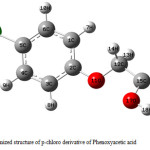 |
Figure 3: Optimized structure of p-chloro derivative of Phenoxyacetic acid.
|
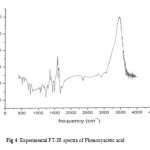 |
Figure 4: Experimental FT-IR spectra of Phenoxyacetic acid.
|
 |
Figure 5: Experimental FT-IR spectra of p-chloro derivative of Phenoxyacetic acid.
|
As there are three rotomers around C3-O12, O12-C13 and around C13-C16, the molecule can exist a variety of confirmations. In all the computations the carbon atom skeleton of benzene moiety and oxyacetic acid both were found to be planar. A general observation from comparing the calculated and available X-ray structural parameter is that the most of HF bond lengths are slightly shorter than the experimental data. This may be the result of the neglect of electron correlation by HF theory.
As we can see most of the MP2 and B3LYP bond lengths and angles match with the experimental data, the best agreement between calculated and experimental values is the MP2/6-311++g(d,p) bond lengths and angles.
The optimized bond length of C-C in phenyl ring fall in the range from 1.383 to 1.385Ao (HF), 1.395 to 1.397Ao (B3LYP) and 1.402 to 1.396 Ao for phenoxy acetic acid. The same bond length fall in the range from 1.382 to 1.391Ao (HF), 1.396 to 1.397Ao (B3LYP) and 1.401 to 1.402Ao (MP2) for p-chloro derivative, which are in good agreement with those of experimentally reported values for C-C bond length of phenyl ring of similar molecules [34].
Table 1: Geometrical Parameters for Phenoxyacetic acid.
Bond length(A°) |
|||||
| C1C2 | 1.383 | 1.395 | 1.402 | 1.391 | |
| C2C3 | 1.384 | 1.397 | 1.400 | 1.389 | |
| C3C4 | 1.383 | 1.395 | 1.401 | 1.393 | |
| C4C5 | 1.385 | 1.395 | 1.394 | 1.381 | |
| C5C6 | 1.385 | 1.396 | 1.402 | 1.385 | |
| C6C1 | 1.385 | 1.396 | 1.396 | 1.377 | |
| C3O12 | 1.369 | 1.388 | 1.374 | 1.378 | |
| O12C13 | 1.386 | 1.407 | 1.404 | 1.421 | |
| C13C16 | 1.515 | 1.526 | 1.522 | 1.499 | |
| C16O17 | 1.178 | 1.206 | 1.204 | 1.266 | |
| C16O18 | 1.328 | 1.356 | 1.357 | 1.260 | |
| Bond angle(°) | |||||
| C1C2C3 | 119.6 | 119.4 | 119.2 | 118.9 | |
| C2C3C4 | 120.6 | 120.7 | 120.2 | 120.4 | |
| C3C4C5 | 119.2 | 119.3 | 119.9 | 119.4 | |
| C4C5C6 | 120.5 | 120.4 | 120.4 | 121.0 | |
| C5C6C1 | 119.6 | 119.7 | 119.3 | 119.1 | |
| C2C1C6 | 120.2 | 120.2 | 120.8 | 121.2 | |
| C2C3O12 | 119.6 | 120.3 | 124.5 | 124.4 | |
| C4C3O12 | 119.6 | 118.8 | 115.2 | 115.3 | |
| C3O12C13 | 117.8 | 116.5 | 116.4 | 115.6 | |
| O12C13C16 | 113.2 | 112.4 | 112.1 | 110.9 | |
| C13C16O17 | 126.6 | 126.5 | 125.8 | 121.1 | |
| C13C16O18 | 109.9 | 109.8 | 109.6 | 114.8 | |
| O17C16O18 | 123.3 | 123.5 | 124.4 | 124.1 | |
Vibrational Spectral Assignments
In the present study the vibrational assignments are based on the scaled B3LYP/6-311++G(d,p) frequencies, infrared intensities, as well as characteristic “group frequencies” from similar molecules. The 51 normal modes of phenoxyacetic acid and p-chloro derivative of phenoxyacetic acid are distributed amongst the symmetry species at Г3N-6=35A'(in plane)+16A”(out of plane) in agreement with Cs symmetry. The detailed vibrational assignment of fundamental modes of phenoxy acetic acid and p-chloro derivative of phenoxyacetic acid along with the experimental and calculated frequencies, IR intensities and normal mode description are reported in table 3 and 4. For visual comparison the experimental and calculated FT-IR spectra of phenoxyacetic acid and p-chloro derivative of phenoxyacetic acid are presented in figure 1 and 2 respectively. Comparison of the frequency calculated at B3LYP with experimental value reveals the overestimation of the calculated vibrational modes due to neglect of anharmonicity in real system. It is customary to scale down the calculated harmonic frequencies in order to improve the agreement with the experimental.
Table 2Geometrical
| Parameters | HF/6-311++g(d,p) | B3LYP/6-311++g(d.p) | MP2/6-311++g(d.p) | Experimental |
| Bond length(A°) | ||||
| C1C2 | 1.382 | 1.396 | 1.401 | 1.39 |
| C2C3 | 1.392 | 1.399 | 1.402 | 1.40 |
| C3C4 | 1.376 | 1.386 | 1.397 | 1.39 |
| C4C5 | 1.387 | 1.394 | 1.393 | 1.40 |
| C5C6 | 1.373 | 1.386 | 1.393 | 1.39 |
| C1C6 | 1.391 | 1.397 | 1.402 | 1.36 |
| C2O11 | 1.350 | 1.368 | 1.369 | 1.34 |
| O11C12 | 1.386 | 1.408 | 1.412 | 1.42 |
| C12C15 | 1.512 | 1.517 | 1.513 | 1.46 |
| C15O16 | 1.183 | 1.205 | 1.211 | 1.23 |
| C15O17 | 1.314 | 1.340 | 1.343 | 1.33 |
| Bond angles( °) | ||||
| C1C2C3 | 119.7 | 119.7 | 119.9 | 117.3 |
| C2C3C4 | 120.3 | 120.3 | 120.3 | 119.8 |
| C3C4C5 | 119.7 | 119.5 | 119.5 | 122.3 |
| C2C1C6 | 119.7 | 119.8 | 119.6 | 120.8 |
| C1C6C5 | 120.0 | 119.8 | 120.0 | 120.4 |
| C1C2O11 | 124.6 | 125.7 | 124.8 | 126.2 |
| C3C2O11 | 115.5 | 115.8 | 115.2 | 116.1 |
| C4C5Cl19 | 119.6 | 119.6 | 119.6 | _ |
| C6C5Cl19 | 119.9 | 119.7 | 119.7 | _ |
| C2O11C12 | 119.7 | 118.3 | 116.3 | 117.3 |
| O11C12C15 | 111.5 | 111.8 | 109.6 | 109.5 |
| C12C15O16 | 120.8 | 121.5 | 123.1 | 126.1 |
| C12C15O17 | 115.3 | 114.4 | 112.4 | 111.7 |
| O16C15O17 | 123.7 | 124.0 | 124.3 | 122.9 |
Vibrational Spectral Assignments
In the present study the vibrational assignments are based on the scaled B3LYP/6-311++G(d,p) frequencies, infrared intensities, as well as characteristic “group frequencies” from similar molecules. The 51 normal modes of phenoxy acetic acid and p-chloro derivative of phenoxy acetic acid are distributed amongst the symmetry species at Г3N-6=35A'(in plane)+16A”(out of plane) in agreement with Cs symmetry. The detailed vibrational assignment of fundamental modes of phenoxyacetic acid and p-chloro derivative of phenoxyacetic acid along with the experimental and calculated frequencies, IR intensities and normal mode description are reported in table 3 and 4. For visual comparison the experimental and calculated FT-IR spectra of phenoxyacetic acid and p-chloro derivative of phenoxyacetic acid are presented in figure 1 and 2 respectively. Comparison of the frequency calculated at B3LYP with experimental value reveals the overestimation of the calculated vibrational modes due to neglect of anharmonicity in real system. It is customary to scale down the calculated harmonic frequencies in order to improve the agreement with the experimental.
Carboxylic Acid Vibrations
Vibrational assignments of –COOH group is significant because the herbicidal activity of the title compound is mainly due to either the presence of this moiety or a group that is easily converted to it within the plant tissue [35,
36]. Vibrational analysis of carboxylic acid is made on the basis of carbonyl group and hydroxyl group both.
The –OH stretching band is characterised by very broad band appearing in the region 3700-3500 cm-1. Accordingly, in the present study, the broad FT-IR bands at 3688cm-1 has been assigned to –OH stretching vibration for phenoxyacetic acid and 3664cm-1 for p-chloro derivative of phenoxyacetic acid. However the calculated wavenumber shows positive deviation of ~190 cm-1 for phenoxyacetic acid and 158cm-1 for p-chloro derivative of phenoxyacetic acid may be due to presence of strong hydrogen bonding. Carbonyl absorption are sensitive and both the carbon and oxygen atoms of the carbonyl group move during the vibration and they have nearly equal amplitude. Normally carbonyl group vibrations [37, 38] occur with strong intensity in the region 1740-1660cm-1.The strong FT-IR bands at 1752cm-1 for phenoxyacetic acid and at 1742cm-1 for p-chloro derivative are assigned to C=O stretching vibrations. The theoretically computed values of 1771cm-1 for phenoxyacetic acid and 1751cm-1 for p-chloro derivative respectively with strong intensity also coincides with experimental observation. The in-plane and out-of-plane bending vibrations have also been identified and presented in table 3 and 4 for the title compound. The calculated IR bands at 1304, 1284, 1260,1218 cm-1 for phenoxyacetic acid and at 1354, 1303, 1274, 1258 cm-1 for p-chloro derivative have substantial –OH bending character with enhaned intensities resulting from intermolecular hydrogen bonding intractions. The calculated low frequency bands below 400cm-1 are out of range in the measured spetra.
C-C Vibrations
The C-C aromatic stretch known as semicircle stretching predicted at 1591, 1576, 1487cm-1 for phenoxyacetic acid and 1592, 1568, 1494cm-1 for p-chloro derivative were in excellent agreement with experimentally observed FT-IR values at 1580, 1564, 1478cm-1 and 1590, 1572, 1484cm-1 for phenoxyacetic acid and p-chloro derivative of phenoxyacetic acid respectively. The C-C-C in plane bending modes are attributed to 782 and 615cm-1 for phenoxyacetic acid and 824 and 633cm-1 for p-chloro derivative respectively. The C-C-C out of plane bending vibrations for phenoxyacetic acid are attributed to 506 and 421cm-1. The same vibration for p-chloro derivative of phenoxyacetic acid are obsurb at 511 and 422cm-1.
C-H Vibration
The region 3100-3000cm-1 is the characteristic region for the ready identification of C-H stretching vibrations [39]. In this region, the bands are not affected appreciably by the nature of substituents. There is a broad absorption band in the region 2500-3000cm-1 upon which the C-H stretching frequencies are superimposed. Hence the FT-IR bands at 3028cm-1 for phenoxyacetic acid and 3046cm-1 for p-chloro derivative have been designated to C-H stretching vibrations.The C-H in plane bending vibrations assigned at 1023-1084 cm-1 even though found to be contaminated by CH2 deformation vibrations are found in literature [38,39], while the experimental observations are at 1098 cm-1 for phenoxyacetic acid and 1096 cm-1 for p-chloro derivative. The calculated frequencies at 753-969 cm-1 in phenoxyacetic acid and 795-943 cm-1 in p-chloro derivative for the C-H out of plane bending mode correlates well with the recorded FT-IR values of 748-950 cm-1 in phenoxyacetic acid and 790-934 cm-1 in p-chloro derivative.
OCH2 Vibrations
For the assignments of CH2 group frequencies, basically six fundamentals can be associated to each CH2 group, namely, CH2: symmetric stretching; CH2: asymmetric stretching; CH2: scissoring and CH2: rocking which belong to in plane (A’) species vibrations. In addition to that CH2 wagging and CH2 twisting modes of the CH2 group would be expected to be depolarized for out of plane(A”) symmetry species.
A major coincidence of theoretical values with that of experimental evaluation is found in the symmetric and asymmetric stretching vibrations of methylene moiety. The antisymmetric CH2 stretching vibrations are generally observed in the region 3000-2900 cm-1 while the symmetric stretching will appear between 2900 and 2850 cm-1 [40]. The CH2 antisymmetric stretching vibrations was observed in IR spectra at 3020 cm-1 in phenoxy acetic acid and 3010 cm-1 in p-chloro derivative respectively. The symmetric stretching vibration was also identified in IR at 2960 cm-1 in phenoxy acetic acid and 2946 cm-1 in p-chloro derivative respectively. The theoretically compute
OCH2 Vibrations
For the assignments of CH2 group frequencies, basically six fundamentals can be associated to each CH2 group, namely, CH2: symmetric stretching; CH2: asymmetric stretching; CH2: scissoring and CH2: rocking which belong to in plane (A’) species vibrations. In addition to that CH2 wagging and CH2 twisting modes of the CH2 group would be expected to be depolarized for out of plane(A”) symmetry species.
A major coincidence of theoretical values with that of experimental evaluation is found in the symmetric and asymmetric stretching vibrations of methylene moiety. The antisymmetric CH2 stretching vibrations are generally observed in the region 3000-2900 cm-1 while the symmetric stretching will appear between 2900 and 2850 cm-1 [40]. The CH2 antisymmetric stretching vibrations was observed in IR spectra at 3020 cm-1 in phenoxy acetic acid and 3010 cm-1 in p-chloro derivative respectively. The symmetric stretching vibration was also identified in IR at 2960 cm-1 in phenoxy acetic acid and 2946 cm-1 in p-chloro derivative respectively. The theoretically computed
C-Cl Vibrations
The vibrations belonging to the bond between the ring and the halogen atoms are worth to discuss here, since mixing of several vibrations are possible due to the lowering of the molecular symmetry and the presence of heavy atoms on the periphery of the molecule [41].
The assignment of C-Cl stretching and deformation vibrations have been made by comparison with similar molecules, p-bromophenol [42] and halogen substituted benzene derivatives [43].The C-Cl stretching vibration of substituted phenoxyacetic acids were observed around at 720 cm-1 [19]. The FT-IR band at 658 cm-1 in p-chloro derivative of phenoxyacetic acid assigned to C-Cl stretching vibration. The theoretical wavenumber of C-Cl stretching vibration at 668 cm-1 coincide very well with experimental values. The C-Cl in plane bending and out of plane bending vibration are assigned at 355 cm-1 and 324 cm-1. These assignments are in agreement with literature data [43-46].
Conclusion
We have carried out density and ab initio calculations on the structure and vibrational spectrum of phenoxyacetic acid and p-chloro derivative. Comparison between the calculated and experimental structural parameters indicate that MP2 method is in good agreement with experimentl ones. Vibrational frequencies and infrared intensities calculated by B3LYP/6-311++G(d,p) method agree very well with experimental results. On the basis of agreement between the calculated and observed results, assignments of fundamental vibrational modes of phenoxyacetic acid and p-chloro derivative were examined. The assignments made higher basis set with only reasonable deviations from the experimental values, seems to be correct. This study demonstrate that scaled DFT/B3LYP calculations are powerful approach for understanding the vibrational spectra of medium sized organic compound.
Reference
- R. Gurumurthy, K. Sathyanarayana, M. Gopalkrishnan, Bull. Chem. Soc., Jpn., 65, 1096(1992).
- B. Karthikeyan, R. Gurumurthy, M. Gopalakrishnan, M. Selvaraju, Asian Chem. ,Lett. 2-3, 97(1998).
- T. Csrhati, E. Forgacs, J.Chromatogr. B: Biomed, Sci. Appl., 717 (1-2) 157(1998).
- A. S. Crafts, Adv. Pest Control Res., 1, 39(1957).
- S. Sarac, C. Safak, H. Erdogan, U. Abbasoglu, Y. Gunay, Eczacilik Fak. Derg. 11 (1) (1991) 1.
- M. Negwar, Organic Chemical Drugs and their Synonyms, Vol , Academic Verlag, Berlin 1996.
- H. Marlin, The Scientific Principles of Crop Protection, Arnold, London, 1973.
- E.B. Wilson, J.C. Decius, P.C. Cross, Molecular Vibration, Mc. Graw Hill, Newyork, 1955.
- G. M .Barrow, Introduction to Molecular Spectroscopy, Mc-Graw Hill Book Co. , Tokyo (1962).
- C. N .Banwell and E.M. Mc. Cash, Fundamentals of Molecular Spectroscopy, Tata Mc Graw. Hill, Publication Co, Ltd. New Delhi, Sixth reprint (1999).
- J. Bina, C. James, I. Hubert, Joe, V. S .Jayakumar, J. Mol. Struct. , 32, 784(2006).
- C. James, A .Amal Raj, R. Raghunathan, V. S. Jayakumar, I. Hubert Joe, J. Raman Spectroscopy, 37, 1381(2006).
- R. F. Liu, R. T. Dennis, A. C. Jeffery, R. M. Paula, J. Phys. Chem. , 100, 3430(1996).
- X. F. Zhou, R. F. Liu, Spectrochim Acta, 53 A, 259(1997).
- O. Nwobi, J. Higgins, X. F. Zhou, R. F. Liu, Chem. Phys. , Lett 272, 155(1997).
- Z. Y. Zhou, D. M. Du, A. P. Fu, Vibr. Spectro., 23, 1951(2000).
- O. Tichcheno, E. S. Kryachko, M. T. Nguven, Spectrochim, Acta A 58, 1951(2002).
- V. Ashok Babu, G. Ramana Rao, Ind. J. Pure Applied Phys. , 25, 203(1987).
- V. Ashok Babu, G. Ramana Rao, Ind. J. Pure Applied Phys. , 25, 209(1987).
- V. Ashok Babu, G. Ramana Rao, Ind. J. Pure Applied Phys. , 25, 216(1987).
- V. Ashok Babu, G. Ramana Rao, Ind. J. Pure Applied Phys. ,28, 1 (1990).
- Colin H. L. Kennard, Graham Smith, Allan H. White, Acta Cryst B, 38, 868-875(1982).
- S. Vijaykumar, L. M. Rao, Acta Crystallogr B, 38, 2062(1982).
- B. Karthikeyan, B. Saravanan, Spectrochim Acta A, 63 (3), 619(2006).
- N .Sundaraganeshan, C. Meghanathan, B.Karthikeyan, Spectrochim Acta Part A, 70, 430-438(2008).
- C. F. Koelsch, J. Am. Chem. Soc. , 53 304(1931).
- Gaussian 09, Revision A.02, M.J. Frisch, G.W. Trucks, H.B. Schlegel, G.E. Scuseria,
- M.A. Robb, J.R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G.A. Petersson,
- H. Nakatsuji, M. Caricato, X. Li, H.P. Hratchian, A.F. Izmaylov, J. Bloino, G. Zheng,
- J.L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida,
- T. Nakajima,Y. Honda, O. Kitao, H. Nakai, T. Vreven, J.A. Montgomery Jr., J.E. Peralta,
- F. Ogliaro, M. Bearpark, J.J. Heyd, E. Brothers, K.N. Kudin, V.N. Staroverov,
- R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J.C. Burant, S.S. Iyengar, J.
- Tomasi, M. Cossi, N. Rega, J.M. Millam, M. Klene, J.E. Knox, J.B. Cross, V. Bakken,
- C. Adamo, J. Jaramillo, R. Gomperts, R.E. Stratmann, O. Yazyev, A.J. Austin, R.
- Cammi, C. Pomelli, J.W. Ochterski, R.L. Martin, K. Morokuma, V.G. Zakrzewski,
- G.A. Voth, P. Salvador, J.J. Dannenberg, S. Dapprich, A.D. Daniels, O. Farkas, J.B.
- Foresman, J.V. Ortiz, J. Cioslowski, D.J. Fox, Gaussian, Inc., Wallingford, CT, 2009.
- P .Hohenberg, W. Kohn, Phys. Rev. 136, B 864(1964).
- D. Becke, J .Chem. Phys. , 98, 5648(1993).
- C. Lee, W. Yang, R. G. Parr, Phys. Rev. , B 37 785(1988).
- K. Chandrasekar, P. Vasantha, Acta Cryst. B 33, 1257(1977).
- C. H. L. Kennard, G. Smith, Acta Cryst B 37, 1454(1981).
- S. V. Kumar, L. M. Rao, Acta Cryst B 38, 2062(1982).
- M. Kurt, M. Yurdakul, S. Yurdakul, J. Mol. Struature (Thiochem), 711, 25(2004).
- R. Cremlyn, Pesticides, Wiley, New York, 1979.
- G. S. Gruzdyev, V. A. Zinchenko, V. A. Kalinin, R. I. Slovtsov, The Chemical Protection of Plants, Mir. Pub. , Moscow, 1983.
- R. L. Peesole, L. D. Shields, I. C. Cairus, M. C. Willam, Modern methods of chemical analysis, Wiley, New York, 1976.
- T. Chithambarathanu, V. Umayourbagan, V. Krishnakumar, Ind. J. Pure Appl. Phys. 40, 72(2002).
- G. Varsanyi, Assignments for Vibrational Spectra of Seven Hundred Benzene derivatives, vols and Academic Kiaclo, Budapest. 1973.
- V. Krishnakumar, R. John Xavier, T. Chithambarathanu, Spectrochim Acta 62 A, 931(2005).
- M. Bakiler, I. V. Maslov, S. Akyuz, J. Mol. Structure, 475, 83(1999).
- W. Zierkiewiez, D.Michalsa, Th Zeegers-Huyskens, J. Phys. Chem. 104 A, 11685(2000).
- G. Varsanyi, Assignments for Vibrational Spectra of Seven Hundred Benzene derivatives, vols and Adam Hilger, 1974.
- E. F. Mooney, Spetrochim Acta, 20, 1021(1964).
- E .F. Mooney, Spetrochim Acta, 19, 877(1963).
- M. Alcolea, Palafox, Int. J. Quantum Chem. 77, 661(2000).

This work is licensed under a Creative Commons Attribution 4.0 International License.
![]()
A New Edition of Web of Science
Journal Impact Factor
2022: 0.5
Five Year: 0.8
Journal is Indexed in

Cabells Whitelist
![]()


