Computational study of N-(2,4-Dichlorophenyl)benzamide
C. Y. Panicker, H. T. Varghese, Y. S. Mary, G.Krishnakumar, P. S. Amala Devi and B. Harikumar, K. Raju
1Department of Physics, TKM College of Arts and Science, Kollam - 691005 (India).
2Department of Physics, Fatima Mata National College, Kollam - 691001 (India).
3Department of Physics, Univeristy College, Thiruvananthapuram (India).
4Department of Physics, College of Engineering, Thiruvananthapuram (India).
5Department of Physics, S.N.College, Chempazhanthy, Thiruvananthapuram, (India).
6Department of Chemistry, TKM College of Arts and Science, Kollam - 691 005 (India).
Article Received on :
Article Accepted on :
Article Published : 02 Mar 2010
The vibrational wavenumbers of N-(2,4-Dichlorophenyl)benzamide were calculated using Gaussian03 software package and the fundamental modes are assigned. The predicted infrared, Raman activities, force constants and polarization rations are reported. The first hyperpolarizability is calculated and the N-(2,4-Dichlorophenyl) benzamide is an attractive object for future studies of non linear optics. The calculated geometrical parameters are in agreement with the reported experimental values.
KEYWORDS:N-(2,4-Dichlorophenyl) Benzamide; Benzamide; HF Calculations; Hyperpolarizability
Download this article as:| Copy the following to cite this article: Panicker C. Y, Varghese H. T, Mary Y. S, Krishnakumar G, Devi P. S. A, Harikumar B, Raju K. Computational study of N-(2,4-Dichlorophenyl)benzamide. Orient J Chem 2010;26(1). |
| Copy the following to cite this URL: Panicker C. Y, Varghese H. T, Mary Y. S, Krishnakumar G, Devi P. S. A, Harikumar B, Raju K. Computational study of N-(2,4-Dichlorophenyl)benzamide. Available from: http://www.orientjchem.org/?p=11508 |
Introduction
Benzamide derivatives exhibit various types of biological properties such as anthelmentic, antihistaminic, antifungal, and antibacterial1-9. Arslan et al.10 reported the molecular structure and vibrational spectra of 2-chloro-N-(diethyl carbamothioyl) benzamide by Hartree-Fock and density functional methods. Takeuchi et al.11 reported the molecular structure of benzamide as studied by gas phase electron diffraction. In the past few decades, the dramatically rising prevalence of multidrug-resistant microbial infections has become a serious health care problem. In particular, the emergence of multi-drug resistant strains of Gram-positive bacterial pathogens such as methicillin-resistant Staphylococcus aureus and Staphylococcus epidermis and vancomycin-resistant Enterococcus is a problem of ever-increasing significance12-16. 6-N-(2-hydroxy-3,5-dichlorophenyl)-2-hydroxy-3,5,6-trichlorobenzamide (oxyclozanide) which has a benzamide structure, was discovered in 1969 as an anthelmentic agent effective against Fasciola hepatica for the treatment of liver fluke infection1. 3,4-Dihydroxy-6-(N-ethylamino)benzamide is a natural product that has been found in green pepper (Piper nigrum L.) as an antibacterial by Pradhan et al.,6. Additionally, a benzamide derivative, BAS-118, has been found to be a novel anti-Helicobacter pylori agent with a potent and selective antibacterial activity, which includes clarithromycin (CAM)- and metronidazole (MNDZ)-resistant isolates6.Yalcin et al.,5,7-9 reported some novel microbiologically active N-(2–hydroxy-5-substitutedphenyl) benzamide/phenyl acetamide/ phenoxy acetamide / thiophenoxy acetamide derivatives. The synthesis and biological evaluation of N-(2-hydroxy-4(or5) -nitro/aminophenyl) benzamides and phenyl acetamides as antimicrobial agents are reported by Ertan et al.,17. 2-hydroxy-N-phenylbenzamides have been reported as a class of compounds with a wide variety of interesting biological activities, including antimycobacterial and antifungal effects . Gas phase structures of fundamental amides, formamide23 and acetamide24 were determined by electron diffraction and their crystal structures were studied by X-ray and neutron diffraction25-29. The crystal structures of benzamide were determined by X-ray and neutron diffraction30,31. The hydrogen bonding interactions between thioacetamide and several N,N-disubstituted benzamides have been studied using near infrared absorption spectroscopy32. Kawski et al.33 reported the X-Ray, IR investigation and quantum mechanical calculations of 2-hydroxy-benzamides. Gowda et al.34 reported the crystal structure of N-(2,4-Dichlorophenyl)benzamide. In the present work, we have calculated the vibrational frequencies and geometrical parameters of the title compound theoretically using Gaussian03 software package and the geometrical parameters are compared with the results given by Gowda et al.34 and other similar derivatives.
Computational Details
Calculations of the title compound were carried out with Gaussian03 program35 using the HF/6-31G* level of theory to predict the molecular structure and wavenumbers. Molecular geometry was fully optimized by Berny’s optimization algorithm using redundant internal coordinates. Harmonic vibrational wavenumbers were calculated using the analytic second derivatives to confirm the convergence to minimum on the potential surface. The wavenumber values computed theoretically contain known systematic errors due to the negligence of electron correlation36. We therefore, have used the scaling factor value of 0.8929 for HF/ 6-31G* basis set. The absence of imaginary wavenumber on the calculated vibrational spectrum confirms that the structure deduced corresponds to minimum energy. The geometrical parameters corresponding to the optimized geometry is given in table 1 together with reported values.
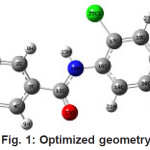 |
Figure 1: Optimized geometry Click here to View figure |
Results and Discussion
The calculated (scaled) wavenumbers and their assignments are given in Table 2. The carbonyl stretching C=O vibrations37,38 are expected in the region 1715-1680 cm-1 and in the present study the calculated value corresponding to this mode appears at 1749 cm. El-Shahawy et al.39 reported a value 1640 cm-1 in the IR spectrum as νC=O for paracetamol. The deviation of the calculated value for this mode can be attributed to the under estimation of the large degree of π-electron delocalization due to conjugation in the molecule40. The NH stretching vibration37 appears as a strong and broad band in the region 3390 ± 60 cm-1. For the title compound, the calculated value for this mode is 3489 cm-1. The CNH vibration, in which the N and H atoms move in opposite direction of the carbon atom in the amide moiety appears at 1528 cm-1 and the CNH vibration, in which the N and H atoms move in the same direction of the carbon atom in the amide group appears at 1253 cm-1. The calculated value of NH rocking is 1097 cm-1. The band at 828 cm-1 is assigned as out-of-plane wagging mode of NH.
For simple organic molecules, CCl stretching vibrations are expected in the region 750-cm-1. Sundaraganesan et al.41 reported õCCl at (IR), 705 (Raman) and at 715 cm-1 theoretically. In the following discussion, the mono and tri-substituted phenyl rings are designated as PhI and PhII, respectively. The benzene ring possesses six ring stretching vibrations of which the four with the highest wavenumbers occurring near 1600, 1580, 1490 and 1440 cm-1 are good group vibrations37. With heavy substituents, the bands tend to shift to somewhat lower wavenumbers, and the greater the number of substituents on the ring, the broader the absorption region37. In the case of C=O substitution, the band near 1490 cm-1 can be very weak37. The fifth ring stretching vibration is active near 1315 ± cm-1, a region that overlaps strongly with that of the CH in-plane deformation37. The sixth ring stretching vibration, the ring breathing mode, appears as a weak band near 1000 cm-1 in mono, 1,3-diand 1,3,5-trisubstituted benzenes. In the otherwise substituted benzenes, however, this vibration is substituent sensitive and difficult to distinguish from other modes.
Table 1: Geometrical Parameters
| Bond lengths(Å) | HF | XRDa | Bond angles (º) | HF | XRDa | Dihedral angles (º) | HF | XRDa |
| C1-C2 | 1.3851 | 1.3874 | A(2,1,6) | 120.0 | 120.1 | D(6,1,2,3) | -0.3 | -0.2 |
| C1-C6 | 1.3847 | 1.3665 | A(2,1,7) | 119.8 | 119.9 | D(2,1,6,5) | 0.7 | 0.5 |
| C1-H7 | 1.0748 | 0.9300 | A(6,1,7) | 120.2 | 119.9 | D(1,2,3,4) | -0.8 | -0.8 |
| C2-C3 | 1.3905 | 1.3753 | A(1,2,3) | 120.2 | 119.7 | D(1,2,3,12) | -179.1 | 179.4 |
| C2-H8 | 1.0752 | 0.9300 | A(1,2,8) | 118.9 | 120.2 | D(2,3,4,5) | 1.4 | 1.6 |
| C3-C4 | 1.3906 | 1.3713 | A(3,2,8) | 120.8 | 120.2 | D(12,3,4,5) | 179.8 | 179.8 |
| C3-C12 | 1.5011 | 1.4883 | A(2,3,4) | 119.5 | 119.3 | D(2,3,12,13) | 152.0 | 146.7 |
| C4-C5 | 1.3826 | 1.3874 | A(2,3,12) | 123.3 | 122.3 | D(2,3,12,14) | -28.9 | -33.6 |
| C4-H9 | 1.0731 | 0.9300 | A(4,3,12) | 117.3 | 118.4 | D(4,3,12,13) | -26.4 | -31.9 |
| C5-C6 | 1.3868 | 1.3564 | A(3,4,5) | 120.3 | 120.9 | D(4,3,12,14) | 152.7 | 147.8 |
| C5-H10 | 1.0749 | 0.9300 | A(3,4,9) | 119.0 | 119.6 | D(3,4,5,6) | -0.9 | -1.3 |
| C6-H11 | 1.0752 | 0.9300 | A(5,4,9) | 120.8 | 119.6 | D(4,5,6,1) | -0.1 | 0.2 |
| C12-O13 | 1.1985 | 1.2212 | A(4,5,6) | 120.0 | 119.2 | D(3,12,14,16) | 178.4 | 176.4 |
| C12-N14 | 1.3681 | 1.3483 | A(4,5,10) | 119.9 | 120.4 | D(13,12,14,16) | -2.6 | -3.9 |
| N14-H15 | 0.9926 | 0.8052 | A(6,5,10) | 120.1 | 120.4 | D(12,14,16,17) | 175.6 | 145.5 |
| C16-N14 | 1.3990 | 1.4083 | A(1,6,5) | 120.0 | 120.8 | D(12,14,16,18) | -5.2 | 35.3 |
| C16-C17 | 1.3951 | 1.3803 | A(1,6,11) | 120.0 | 119.6 | D(14,16,17,19) | 179.4 | 179.8 |
| C16-C18 | 1.3948 | 1.3883 | A(5,6,11) | 120.0 | 119.6 | D(14,16,17,25) | -0.6 | 1.0 |
| C17-C19 | 1.3814 | 1.3743 | A(3,12,13) | 121.3 | 121.6 | D(18,16,17,19) | 0.1 | -1.0 |
| C17-Cl25 | 1.7500 | 1.7352 | A(3,12,14) | 114.9 | 115.9 | D(18,16,17,25) | -179.8 | -179.7 |
| C18-C20 | 1.3814 | 1.3743 | A(13,12,14) | 123.9 | 122.5 | D(14,16,18,20) | -179.3 | -179.2 |
| C18-H21 | 1.0676 | 0.9300 | A(12,14,15) | 116.1 | 119.5 | D(17,16,18,20) | -0.1 | 1.6 |
| C19-C22 | 1.3785 | 1.3743 | A(12,14,16) | 128.4 | 126.5 | D(16,17,19,22) | -0.1 | -0.6 |
| C19-H23 | 1.0719 | 0.9300 | A(15,14,16) | 115.2 | 114.0 | D(25,17,19,22) | 179.8 | 178.1 |
| C20-C22 | 1.3811 | 1.3663 | A(14,16,17) | 119.1 | 120.1 | D(16,18,20,22) | -0.0 | -0.5 |
| C20-H24 | 1.0732 | 0.9300 | A(14,16,18) | 123.6 | 122.2 | D(17,19,22,20) | -0.0 | 1.7 |
| C22-Cl26 | 1.7408 | 1.7432 | A(17,16,18) | 117.4 | 117.8 | D(17,19,22,26) | 180.0 | -177.5 |
| A(16,17,19) | 122.1 | 122.2 | D(18,20,22,19) | 0.1 | -1.2 | |||
| A(16,17,25) | 120.6 | 119.2 | D(18,20,22,26) | -179.9 | 178.1 | |||
| A(19,17,25) | 117.4 | 118.6 | ||||||
| A(16,18,20) | 120.9 | 120.9 | ||||||
| A(16,18,21) | 119.3 | 119.5 | ||||||
| A(20,18,21) | 119.9 | 119.5 | ||||||
| A(17,19,22) | 119.2 | 118.1 | ||||||
| A(17,19,23) | 119.9 | 120.9 | ||||||
| A(22,19,23) | 120.9 | 120.9 | ||||||
| A(18,20,22) | 120.3 | 119.4 | ||||||
| A(18,20,24) | 119.6 | 120.3 | ||||||
| A(22,20,24) | 120.1 | 120.3 | ||||||
| A(19,22,20) | 120.2 | 121.6 | ||||||
| A(19,22,26) | 119.6 | 119.1 | ||||||
| A(20,22,26) | 120.2 | 119.3 |
aXRD data from reference 34
Table 2: Calculated scaled wave numbers and assignments
|
υ (cm-1) |
IR intensity |
Raman activity |
Force constants |
Depolarization ratio (P) |
Depolarization ratio (U) |
Assignments |
|
3489 |
45.05 |
55.02 |
9.7 |
0.16 |
0.27 |
υNH |
|
3103 |
7.7 |
58.43 |
7.78 |
0.2 |
0.33 |
υCH II |
|
3053 |
0.1 |
65.64 |
7.54 |
0.28 |
0.44 |
υCH II |
|
3046 |
4.32 |
99.15 |
7.51 |
0.15 |
0.27 |
υCH I |
|
3039 |
2.76 |
89.49 |
7.46 |
0.33 |
0.49 |
υCH II |
|
3029 |
21.84 |
222.36 |
7.45 |
0.12 |
0.21 |
υCH I |
|
3020 |
31.14 |
58.03 |
7.37 |
0.73 |
0.84 |
υCH I |
|
3010 |
7.84 |
106.49 |
7.3 |
0.7 |
0.82 |
υCH I |
|
3002 |
1.89 |
37.74 |
7.24 |
0.72 |
0.84 |
υCH I |
|
1749 |
262.03 |
94.22 |
20.04 |
0.23 |
0.37 |
υCO |
|
1616 |
33.51 |
86.35 |
9.77 |
0.51 |
0.67 |
υPh I |
|
1606 |
84.77 |
173.67 |
10.05 |
0.44 |
0.61 |
υPh II |
|
1597 |
172.2 |
14.31 |
6.45 |
0.71 |
0.83 |
υPh II |
|
1591 |
25.87 |
2.57 |
9.05 |
0.48 |
0.65 |
υPh I |
|
1528 |
615.55 |
11.62 |
4.55 |
0.55 |
0.71 |
δNH |
|
1491 |
49.37 |
0.17 |
3.61 |
0.6 |
0.75 |
υPh I |
|
1467 |
46.85 |
15.69 |
3.68 |
0.33 |
0.5 |
υPh II |
|
1440 |
7.44 |
4.48 |
3.25 |
0.4 |
0.57 |
υPh I |
|
1379 |
105.49 |
12.33 |
4.52 |
0.3 |
0.46 |
υPh II |
|
1317 |
0.63 |
0.84 |
1.71 |
0.68 |
0.81 |
υPh I |
|
1282 |
411.29 |
72.68 |
2.5 |
0.22 |
0.37 |
υPh II, δCH I |
|
1253 |
146.13 |
14.33 |
2.26 |
0.2 |
0.33 |
δNH, δCH II |
|
1225 |
0.49 |
5.69 |
2.85 |
0.34 |
0.5 |
υCN |
|
1200 |
8.65 |
1.75 |
1.82 |
0.38 |
0.55 |
υPh I |
|
1166 |
10.87 |
3.28 |
1.167 |
0.39 |
0.56 |
δCH I |
|
1149 |
4.43 |
9.2 |
1.53 |
0.18 |
0.31 |
δCH I |
|
1102 |
1.29 |
8.96 |
2.04 |
0.48 |
0.65 |
δCH II |
|
1097 |
11.69 |
13.94 |
2.44 |
0.26 |
0.42 |
δCH II |
|
1085 |
67.44 |
13.18 |
3.59 |
0.58 |
0.73 |
υPh II |
|
1079 |
26.31 |
7.15 |
3.01 |
0.25 |
0.41 |
ρNH |
|
1061 |
6.51 |
0.57 |
1.46 |
0.54 |
0.7 |
δCH I |
|
1031 |
17.87 |
2.23 |
2.91 |
0.38 |
0.55 |
δCH I |
|
1015 |
0.09 |
1.47 |
1.04 |
0.21 |
0.34 |
γCH I |
|
1007 |
6.09 |
15.33 |
1.86 |
0.12 |
0.21 |
υPh I |
|
993 |
0.73 |
0.06 |
1 |
0.55 |
0.71 |
γCH I |
|
993 |
2.46 |
0.95 |
0.97 |
0.75 |
0.85 |
γCH II |
|
974 |
1.65 |
36.74 |
4.21 |
0.12 |
0.21 |
Ring breath I |
|
951 |
1 |
1.597 |
0.95 |
0.7 |
0.82 |
γCH I |
|
906 |
12.91 |
1.67 |
0.85 |
0.75 |
0.86 |
γCH I |
|
877 |
30.22 |
10.4 |
3.71 |
0.11 |
0.19 |
δCO, υNH |
|
860 |
0.07 |
3.43 |
0.69 |
0.41 |
0.58 |
γCH II |
|
854 |
48.06 |
1.06 |
0.76 |
0.71 |
0.83 |
γCH I |
|
828 |
31.73 |
8.59 |
2.89 |
0.17 |
0.29 |
Ring breath II |
|
797 |
1.72 |
0.52 |
1.37 |
0.67 |
0.8 |
γCH II |
|
732 |
39.21 |
1.03 |
2.93 |
0.72 |
0.84 |
δPh(X)I, υCCl |
|
718 |
89.58 |
3.78 |
0.8 |
0.72 |
0.84 |
γCH I, υCCl |
|
710 |
14.52 |
0.67 |
1.11 |
0.69 |
0.82 |
γCH I |
|
681 |
4.59 |
1.14 |
1.06 |
0.24 |
0.38 |
γPh I |
|
664 |
16.21 |
11.03 |
2.2 |
0.15 |
0.26 |
δPh I, II |
|
632 |
2.4 |
3.09 |
2.06 |
0.42 |
0.59 |
δPh I, II |
|
605 |
0.91 |
6.74 |
1.68 |
0.74 |
0.85 |
δPh I |
|
562 |
0.12 |
0.12 |
0.83 |
0.75 |
0.86 |
γPh II |
|
541 |
91.45 |
91.5 |
0.26 |
0.74 |
0.85 |
γPh(X) II |
|
502 |
7.55 |
7.55 |
0.95 |
0.55 |
0.71 |
γPh(X) I |
|
459 |
5.7 |
2.14 |
1.02 |
0.35 |
0.52 |
δPh(X) I |
|
445 |
5.51 |
0.45 |
0.51 |
0.73 |
0.84 |
δPh(X) II |
|
421 |
17.63 |
1.78 |
0.68 |
0.75 |
0.85 |
δPh(X) II |
|
407 |
0.12 |
0.14 |
0.36 |
0.71 |
0.83 |
γPh I |
|
396 |
2.14 |
6.53 |
1.02 |
0.22 |
0.36 |
δPh(X) I |
|
378 |
3.98 |
4.03 |
0.96 |
0.23 |
0.38 |
δCX(X) I |
|
351 |
0.89 |
1.08 |
0.57 |
0.75 |
0.86 |
δCX’(X) II |
|
300 |
11.42 |
0.97 |
0.57 |
0.57 |
0.72 |
γCX(X) II |
|
231 |
0.13 |
3.61 |
0.36 |
0.27 |
0.42 |
δCX’(X) I |
|
209 |
2.16 |
3.18 |
0.23 |
0.74 |
0.85 |
δCX’(X) II |
|
181 |
0.92 |
2.21 |
0.25 |
0.72 |
0.84 |
δCX’(X) II |
|
171 |
0.05 |
1.66 |
0.13 |
0.75 |
0.86 |
δCX(X) I |
|
152 |
2.02 |
3.46 |
0.14 |
0.68 |
0.81 |
γCX’(X) II |
|
123 |
0.78 |
1.74 |
0.06 |
0.74 |
0.85 |
γPh(X) I,II |
|
74 |
2.41 |
1.45 |
0.03 |
0.72 |
0.83 |
γPh(X) I |
|
51 |
2.48 |
3.48 |
0.01 |
0.74 |
0.85 |
γPh(X) I |
|
29 |
0.04 |
0.91 |
0.01 |
0.75 |
0.86 |
γPh(X) II |
|
16 |
2.89 |
3.65 |
0.01 |
0.75 |
0.86 |
γPh(X) I |
υ-stretching; δ- in-plane deformation; γ- out-of-plane deformation; ρ -rocking; ω-waging; PhI- mono substituted phenyl ring; PhII-trisubstituted phenyl ring; X-substituent sensitive
In asymmetric trisubstituted benzenes, when all the three substituents and light, the wavenumber interval of the breathing mode42 is between 500 and 600 cm-1. When all the three substituents are heavy, the wavenumber appears above 1100 cm-1. In the case of mixed substituents42, the wavenumber is expected to appear between 600 and 750 cm-1. The in-plane and out-of-plane CH deformations37 of the phenyl ring are expected above 1000 cm and below 1000 cm-1 and these modes are assigned (Table 2).
At C3 position, the angle C4-C3-C12 is reduced by 2.7º and C2-C3-C12 is increased by 3.3º from 120º, and this asymmetry of exocyclic angles reveals the interaction between O13 and the phenyl ring I. The C-C bond lengths in the phenyl ring lie between 1.3564 to 1.3874Å whereas for the phenyl ring II, the range is 1.3663 to 1.3883Å. The CH bond lengths lie between 1.0731 and 1.0752Å for ring I and between 1.0676 and 1.0732Å for ring II. According to Noveron el al.43 for complexes of N-(4-pyridyl)benzamide, the bond lengths for C16-N14, C12-O13, C12-N14, C12-C3, C3-C2, C3-C4 and N14-H15 are 1.3953, 1.2253, 1.3703, 1.4943, 1.3923, 1.3933 and 0.773Å, respectively and for the title compound the corresponding bond values are 1.399, 1.1985, 1.3681, 1.5011, 1.3905, 1.3906 and 0.9926Å, respectively. For N-(2-pyridyl)benzamide complexe44, the bond lengths for C12-O13, N14-C12, N14-C16 and C12-C3 are 1.2445, 1.3646, 1.4156 and 1.4810Å, respectively and in the present case, the corresponding values are 1.1985, 1.3681, 1.399 and 1.5011Å. The C=O and C-N bond lengths11 in benzamide, acetamide and formamide are, respectively, 1.2253, 1.2203, 1.2123 and 1.3801, 1.3904, 1.3683Å. According to literature25,27,31,45 the changes in bond lengths in C=O and C-N are consistent with the following interpretation: that is hydrogen bond decreases the double bond character of C=O bond and increases the double bond character of C-N bond. The values of the angles C4-C3-C12 (117.3º) and C3-C12-O13 (121.3º) are smaller than those of benzaldehyde46, 121.0º and 123.6º. These differences are due to the steric repulsion between H8 and H15 atoms.
The dihedral angles C2-C3-C12-O13 was determined to be 152.0º by the HF method. On the contrary, the equilibrium structure of benzaldehyde46 is planar. The steric repulsion in the present case is also considered to cause the nonplanar skeleton. The C3-C12 bond length (1.5011) is larger than the corresponding length of benzaldehyde46 (1.4794) by 0.0217. Saeed et al.47 reported C12-O13, N14-C12 and N14-C16 bond lengths as 1.2132, 1.3612 and 1.4042 for benzamide derivatives. For the title compound, the bond lengths obtained are 1.1985, 1.3681 and 1.399. For the benzamide moiety of the title compound, the calculated values of the bond angles C12-N14-C16, C18-C16-N14, O13-C12-N14, O13-C12-C3, N14-C12-C3, C4-C3-C12 and C2-C3-C12 are 128.4, 123.6, 123.9, 121.3, 114.9, 117.3 and 123.3º, respectively. Sun et al.48 reported the corresponding angles as 124.7, 116.6, 123.1, 118.7, 118.3, 115.6 and 124.3º, respectively. Also, the HF calculations give the torsional angles C19-C17-C16-N14, N14-C16-C19-C20, O13-C12-C3-C2, N14-C12-C3-C2, O13-C12-C3-C4 and N14-C13-C3-C4 as 177.4, -179.3, 152.0, -28.9, – 26.4 and 152.7º around the benzamide group, which are in agreement with the reported values43 178.5, -178.9, -16.8, 163.8, 161.6 and -17.8º. The C12-N14 bond is twisted from the phenyl ring I and II as is evident from the torsion angles C18-C16-N14-C12 = -5.2º, C17-C16-N14-C12 = 175.6º and N14-C12-C3-C4 = 152.7º, N14-C12-C3-C2 = -28.9º.
Analysis of organic molecules having conjugated π-electron systems and large hyperpolarizability using infrared and Raman spectroscopy has evolved as a subject of research49. The potential application of the title compound in the field of non linear optics demands the investigation of its structural and bonding features contributing to the hyperpolarizability enhancement, by analyzing the vibrational modes using the IR and Raman spectrum. The first hyperpolarizability (β0) of this novel molecular system is calculated using HF method, based on the finite field approach. In the presence of an applied electric field, the energy of a system is a function of the electric field. First hyperpolarizability is a third rank tensor that can be described by a 3×3×3 matrix. The 27 components of the 3D matrix can be reduced to 10 components due to the Kleinman symmetry50. The calculated first hyperpolarizability of the title compound is 1.610-30 esu, which is comparable with the reported values of similar derivatives51 and experimental evaluation of this data is not readily available. We conclude that the title compound is an attractive object for future studies of non linear optical properties.
References
- Mrozik, H., Jones, H., Friedman, J., Schwartzkopf, G., Schardt, R., Patchett, A., Holff, D.R., Yakstis, J.J., Riek, R.F., Ostlind, D.A., Plischker, G.A., Butler, R.W., Cuckler, A.C., and Campbell, W.C., Experientia, 25: 883 (1996).
- Japan Patent, 73, 37, 819, Chem. Abstr. 81 (1974) 73387 (1973).
- Braz Pedido, P.I., N80 04, 641, Chem. Abstr. 95 (1981) 61812z (1981).
- White, G.A., Pestic. Biochem. Physiol. 34: 255 (1989).
- Yalcin, I., Kaymakcioglu, B.K., Oren, I., Sener, E., Temiz, O., Akin, A., and Altanlar, N., Il Farmaco., 52: 685 (1997).
- Pradhan, K.J., Variyar, P.S., and Bandekar, J.R., Lebensm. Wiss. U-Technol., 32: 121 (1999).
- Aki-Sener, E., Bingol, K.K., Oren, I., Temiz-Arpaci, O., Yalcin, I., and Altanlar, N., Il Farmaco., 55: 469 (2000).
- Aki-Sener, E., Bingol, K.K., Temiz-Arpaci, O., Yalcin, I., and Altnlar, N., Il Farmaco., 57: 451(2002).
- Yildiz-Oren, I., Aki-Sener, E., Ertas, C., Temiz-Arpaci, O., Yalcin, I., and Altanlar, N., Turk. J. Chem., 28: 441 (2004).
- Arslan, H., Florke, U., Kulcu, N., and Binzet, G., Spectrochim. Acta, 68A: 1347 (2007).
- Takeuchi, H., Sato, M., Tsuji, T., Takashima, H., Egawa, T., and Konaka, S., J. Mol. Struct., 175: 485-486 (1999).
- Dalhoff, A., Infection, 22: 111 (1994).
- Lee, V., and Hecker, S., J. Med. Chem., 19: 521 (1999).
- Livermore, D., Int. J. Antimicrob. Agents 16: 3 (2000).
- 15. Poole, P., Curr. Opin. Microbiob. Agents 4:500 (2001).
- Abbant, D., Macielag, M., and Bush, K., Expert. Opin. Investig. Drugs, 12: 379 (2003).
- Ertan, T., Yildiz, I., Ozkan, S., Temiz-Arpaci, O., Kaynak, F., Yalcin, I., Aki-Sener, E., and Abbasoglu, U., Bioorg. Med. Chem., 15: 2032 (2007).
- Vinsova, J., and Imramovsky, A., Cesk. Slov. Farm., 53: 294 (2004).
- De La Fluente, R., Sonawane, N.D., Arumainayagam, D., and Verkaman, A.S., Br. J. Pharmacol., 149: 551 (2006).
- Vinsova, J., Imramovsky, A., Buchta, V., Ceckova, M., Dolezal, M., Staud, F., Jampilek, J., and Kaustova, J., Molecules, 11: 1 (2007).
- Dahlgren,M.K., Kauppi, A.M., Olsson, I.M., Linusson, A., and Elofsson, M., J. Med. Chem., 50: 6177 (2007).
- Waisser, K., Matyk, J., Divisova, H., Husakova, P. Kunes, J., Klimesova, V., Kaustova, J., Moellmann, U., Cause, H.M, and Miko, M., Arch. Pharm., 149: 616 (2006).
- Kitano, M., and Kuchitsu, K., Bull. Chem. Soc. Jpn., 47: 67 (1974).
- Kitano, M., and Kuchitsu, K., Bull. Chem. Soc. Jpn., 46: 3048 (1973).
- Stevens, E.D., Acta Cryst., B34: 544 (1978).
- Ladell, J., and Post, B., Acta Cryst., 7: 559 (1954).
- Otterson, T., Acta Chem. Scand., A29: 939 (1975).
- Jeffrey, G.A., Ruble, J.R., McMullan, R.K., DeFrees, D.J., Binkley, J.S., and Pople, J.A., Acta Cryst., B36: 2292 (1980).
- Denne, W.A., and Small, R.W.H., Acta Cryst., B27: 1094 (1971) .
- Blake, C.C.F, and Small, R.W.H, Acta Cryst., B28, 2201 (1972).
- Gao, Q., Jeffrey, G.A., Ruble, J.R., and McMullan, R.K., Acta Cryst., B47: 742 (1991).
- Choi, Y.S., Kim, J., Park, J., Yu, J.A., and Yoon, C.J., Spectrochim. Acta, 52A: 1779 (1996).
- Kawski, P., Kochel, A., Perevozkina, M.G., and Filarowski, A., J. Mol. Struct., 790: 65 (2006).
- Gowda, B.T., Tokarick, M., Kozisek, J., Sowmya, B.P., and Fuess, H., Acta Cryst. E64: o950 (2008,).
- Frisch, M.J., et al., Gaussian03, Revision C.02., Gaussian Inc., Wallingford, CT (2004).
- Foresman, J.B., in: Frisch, E., (Ed.), Exploring Chemistry with Electronic Structure Methods: A Guide to Using Gaussian, Pittsburg, PA, (1996).
- Roeges, N.P.G., A Guide to the Complete Interpretation of Infrared Spectra of Organic Structures, Wiley, New York (1994).
- Barthes, M., De Nunzio, G., and Riber, G., Synth. Met. 76: 337 (1996).
- El-Shahawy, A.S., Ahmed, S.M., and Sayed, N.K., Spectrochim. Acta 66: 143 (2007).
- Panicker, C.Y., Varghese, H.Y., Philip, D., Nogueira, H.I.S., and Kastkova, K., Spectrochim. Acta 67: 1313 (2007).
- Sundaraganesan, N., Meganathan, C., Joshua, B.D., Mani, P., and Jayaprakash, A., Spectrochim. Acta 71: 1134 (2008).
- Varsanyi, G., Assignments of Vibrational Spectra of Seven Hundred benzene Derivatives, Wiley, New York (1974).
- Noveron, J.C., Arif, A.M., and Stang, P.J., Chem. Mater., 15: 372 (2003).
- Sun, W.H., Zhang, W., Gao, T., Tang, X., Chen, L., Li, Y., and Jin, X., J. Organomet. Chem. 689: 917 (2004).
- Katz, J.L., and Post, B., Acta Cryst. 13: 624 (1960).
- Borisenko, K.B., Bock, C.W., and Hargittai, I., J. Phys. Chem. 100: 7426 (1996).
- Saeed, A., Hussain, S., and Florke, U., Turk. J. Chem. 32: 1 (2008).
- Lee, S.Y., and Hoo, B.H, Bull. Korean Chem. Soc. 17: 760 (1996).
- Tommasini, M., Castiglioni, C., Del Zoppo, M., and Zerbi, G., J. Mol. Struct. 480: 179 (1999).
- Kleinman, D.A., Phys. Rev., 126: 1977 (1962). 51. Varghese, H.T., Panicker, C.Y., Madhavan, V.S., Mathew, S., Vinsova, J., and Van Alsenoy, C., J. Raman Spectrosc. Doi:10.1002/jrs.2265.

This work is licensed under a Creative Commons Attribution 4.0 International License.
![]()
A New Edition of Web of Science
Journal Impact Factor
2022: 0.5
Five Year: 0.8
Journal is Indexed in

Cabells Whitelist
![]()


