Study of the Stability and Chemical Reactivity of a Series of Tetrazole Pyrimidine Hybrids by the Density Functional Theory Method (DFT)
Ahissandonatien Ehouman1* , Adjoumanirodrigue Kouakou1, Fatogoma Diarrassouba1, Hakim Abdel Aziz Ouattara1 and Paulin Marius Niamien2
, Adjoumanirodrigue Kouakou1, Fatogoma Diarrassouba1, Hakim Abdel Aziz Ouattara1 and Paulin Marius Niamien2
1Laboratoire de Thermodynamique et Physico-Chimie du Milieu, UFR SFA, Université Nangui Abrogoua, 02 BP 801 Abidjan 02, Côte-d’Ivoire.
2Laboratoire de Constitution et Réaction de la Matière, UFR SSMT, Université Félix Houphouët-Boigny, Abidjan Cocody, 22 BP 582 Abidjan 22.
Corresponding Author E-mail: ehoumandona@gmail.com
DOI : http://dx.doi.org/10.13005/ojc/370406
Article Received on : 24 Mar 2021
Article Accepted on : 01 Jul 2021
Article Published : 14 Jul 2021
Reviewed by: Dr. Paul Metilda
Second Review by: Dr. Likaa Khalid
Final Approval by: Dr. Leila Hojat
Our theoretical study of stability and reactivity was carried out on six (06) molecules of a series of pyrimidine tetrazole hybrids (PTH) substituted with H, F, Cl, Br, OCH3 and CH3 atoms and groups of atoms using the density function theory (DFT). Analysis of the thermodynamic formation quantities confirmed the formation and existence of the series of molecules studied. Quantum chemical calculations at the B3LYP / 6-311G (d, p) level of theory determined molecular descriptors. Global reactivity descriptors were also determined and analyzed. Thus, the results showed that the compound PTH_1 is the most stable, and PTH_5 is the most reactive and nucleophilic. Similarly, the compound PTH_4 is the most electrophilic. The analysis of the local descriptors and the boundary molecular orbitals allowed us to identify the preferred atoms for electrophilic and nucleophilic attacks.
KEYWORDS:Chemical Stability; DFT; Global and local descriptors; Reactivity.; Tetrazole Pyrimidine Hybrids (PTH)
Download this article as:| Copy the following to cite this article: Ehouman A, Kouakou A, Diarrassouba F, Ouattara H. A. A, Niamien P. M. Study of the Stability and Chemical Reactivity of a Series of Tetrazole Pyrimidine Hybrids by the Density Functional Theory Method (DFT). Orient J Chem 2021;37(4). |
| Copy the following to cite this URL: Ehouman A, Kouakou A, Diarrassouba F, Ouattara H. A. A, Niamien P. M. Study of the Stability and Chemical Reactivity of a Series of Tetrazole Pyrimidine Hybrids by the Density Functional Theory Method (DFT). Orient J Chem 2021;37(4). Available from: https://bit.ly/3kh74Jx |
Introduction
An infection, defined as an invasion of the organism by a foreign agent, namely a bacterium or a virus that can cause a pathological condition by injury to local cells. Among them, we have the invasive fungal infections (IFI), which can be caused by a variety of fungi such as yeasts, molds (Aspergillusspp) and dimorphic fungi (Histoplasmacapsulatum) that are very common in some immunocompromised patients 1,2. We have more than 300 million people suffering from these diseases causing 1.35 million deaths each year in the world, which poses a real problem to the global health system 2,3. In the face of this scourge, clinically available antifungal agents have been proposed for the treatment of these IFIs but they are quite limited 4,5 and most of them are not effective due to high resistance to toxicity and some adverse side effects 6,7. Therefore, the development of new antifungal agents with low toxicity that will be effective in the control of these fungi are in order. As a result, tetrazole hybrids have been synthesized and tested for their antifungal activities by Sheng-Qiang Wang et al 8,
some of which have shown promising activity against drug-sensitive and drug-resistant fungi. Among these hybrids, we distinguish the pyrimidine associated with tetrazole which will give rise to a new series of molecules, namely Pyrimidine-Tetrazole Hybrids. This study will allow us to consider which of them are the most likely to act as antifungal.
Materials and Methods
This study was performed on six (06) molecules of the Pyrimidine Tetrazole Hybrid series presented in Table 1 below:
 |
Table 1: Structure of the studied Pyrimidine Tetrazole Hybrid derivatives. |
Software and theory level
The theoretical study of stability and chemical reactivity was carried out based on three theoretical approaches. The first approach relates to global descriptors deriving from frontier molecular orbitals and then the second approach is concerned with local indices of reactivity. The geometries of the molecules were optimized at the DFT calculation level with the B3LYP functional 9-11 in the 6-311 G (d, p) base using the Gaussian 09 software 12. This Hybrid functional gives better energies and is in agreement with high level ab initio methods 13-14.
As for the split-valence and triple-dzeta base (6-311G (d, p)), it is sufficiently extensive and the consideration of polarization functions is important for the explanation of free doublets of heteroatoms. The geometries are kept constant for cationic and anionic systems. The global reactivity indices were obtained from the conceptual DFT model 15-17.
Standard thermodynamic quantities of formation
The thermodynamic quantities of the molecules were performed from optimization and calculation of frequencies at the B3LYP / 6-311G (d, p) level. The quantities such as entropy, enthalpy and free enthalpy of formation were determined using the following formulas proposed by Otchersky et al. 11.

∑D0 : Atomization energy
ε0 (M) : Total energy of the molecule
εZPE : Zero point energy of the molecule

Enthalpy corrections of atomic elements. These values are included in the Janaf table 19.

Molecule enthalpy correction
Hcorr : Enthalpy of thermal correction

x : Number of atoms in the Molecule.
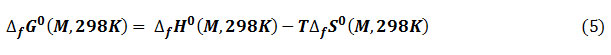
Reactivity descriptors
Global descriptors
To predict chemical reactivity, some theoretical descriptors related to conceptual DFT have been determined. In particular, the energy of the Lowest Unoccupied Molecular Orbital (ELUMO), the energy of the Highest Occupied Molecular Orbital (EHOMO), electronegativity (χ), softness global (σ) and the global electrophilicity index (ω). These descriptors are all determined from the optimized molecules. It should be noted that the descriptors related to the frontier molecular orbitals have been calculated in a very simple way within the framework of the Koopmans approximation 18. LUMO energy characterizes the sensitivity of the molecule to nucleophilic attack, and HOMO energy characterizes the susceptibility of a molecule to electrophilic attack. Electronegativity (χ) is the parameter that reflects the ability of a molecule not to let its electrons escape. The overall softness (σ) expresses the resistance of a system to a change in its number of electrons. The overall electrophilicity index characterizes the electrophilicity of the molecule. These different parameters are calculated from equations (6):
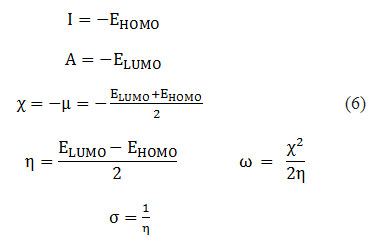
Local Fukui indices
The local responsiveness study was also conducted by determining the local Fukui indices. Two indices were used:
The electrophilic Fukui function f+ which characterizes the electron density response to electron gain and gives information on the most electrophilic site towardnucleophilic attack and the function of nucleophilic Fukui f– characterizing the electron density response to electron loss and provides information on the most nucleophilic site toward an electrophilic attack.
It has been shown [19], for reactions controlled by frontier orbitals, that a high value of the Fukui index means high reactivity of the site. The condensed form of these two functions in the case of a molecule with N electrons has been proposed by Yang and Mortier 20.

Results and Discussion
Study of standard thermodynamic formation parameters
The thermodynamic parameters, namely the enthalpy of formation ΔfH°, the enthalpy of formation ΔfS° and the free enthalpy of formation ΔfG° were also determined. It should be noted that the enthalpy reflects the thermicity of a chemical reaction when entropy provides information on the level of disorder in the system. In addition, the free enthalpy reflects the possibility or not of a chemical reaction. The values of the thermodynamic parameters are given in Table 2.
Table 2: Thermodynamic quantities of PTH formation optimized at the theory level B3LYP / 6-311G (d, p).
|
Molecule |
(kcal/mol) |
(cal/mol.K) |
(kcal/mol) |
|
PTH_1 |
-982.1206 |
-755.3723 |
-756.9064 |
|
PTH_2 |
-1050.5583 |
-761.2891 |
-823.5800 |
|
PTH_3 |
-982.1498 |
-760.0310 |
-755.5466 |
|
PTH_4 |
-971.4305 |
-759.4040 |
-745.0142 |
|
PTH_5 |
-1131.1434 |
-871.9923 |
-871.1589 |
|
PTH_6 |
-1039.4355 |
-837.8549 |
-789.6291 |
The results show that all the values of the standard thermodynamic quantities of molecule formation are negative. This means that during the formation of all the PTH compounds occurs spontaneously with a release of heat and a reduction in disorder. At this level, we note that the quantities determined at the level of theory B3LYP / 6-311G (d, p) confirm the formation and existence of the series of Pyrimidine Tetrazole Hybrids explored.
Global reactivity indices
The conceptual DFT descriptors determined have contributed in several ways to understanding the structure of molecules and their reactivity. The reactivity parameters examined in this series of compounds are: chemical potential (µ), hardness (ɳ), softness (S) and electrophilic index (ω). The calculated values of said parameters are reported in table (3).
Table 3: Summary of global reactivity descriptors at the B3LYP / 6-311G theory level (d, p).
|
Molecule |
µ (eV) |
η (eV) |
S (eV-1) |
ω (eV) |
|
PTH_1 |
5.2518 |
2.1107 |
0.4738 |
6.5338 |
|
PTH_2 |
5.2806 |
2.0671 |
0.4838 |
6.7449 |
|
PTH_3 |
5.3283 |
2.0304 |
0.4925 |
6.9913 |
|
PTH_4 |
5.2838 |
1.9905 |
0.5024 |
7.0127 |
|
PTH_5 |
4.8228 |
1.8757 |
0.5331 |
6.2002 |
|
PTH_6 |
5.0925 |
2.0401 |
0.4902 |
6.3560 |
The chemical potential values recorded for the Pyrimidine Tetrazole Hybrids in Table 3 indicate that PTH_3 is the molecule most likely to retain its electrons due to its fairly high chemical potential value compared to others.
In terms of chemical hardness (η) and overall softness (S), the hardness increases with the stability of the molecule while the softness increases with the reactivity of the molecule. Thus, the harder a molecule, the more stable and less reactive it is. This is the case with PTH_1, which is the hardest molecule, and therefore the least reactive of all, because it has the highest hardness value (η). The PTH_5 molecule, on the other hand, is the softest of all molecules, hence the most reactive. Methoxy therefore contributes to increasing the reactivity of the molecule.
With regard to the electrophilic index (ω), a molecule is all the more electrophilic the higher the value of the overall electrophilic index, the molecule exhibiting the greater value of the electrophilic index is PTH_4 while the compound PTH_5 is the least electrophilic because it has the lowest value. Considering the other values in the table, we deduce that the halogens contribute to the increase in the electrophilic character of our molecules while the electron-donor groups decrease this character.
Local descriptors
Since the dimerization of a biologically active molecule is likely to result in a biologically active molecule, we have therefore found it necessary to search for sites of nucleophilic and electrophilic attacks.
The Fukui indices, the frontier molecular orbitals HOMO and LUMO as well as the coefficients of the atomic orbitals in the frontier orbitals characterize the sites of electrophilic and nucleophilic attacks located for each molecule studied.
Visualization of frontier molecular orbitals
The analysis of the frontier molecular orbitals of PTH_1 is given in figure 1 below
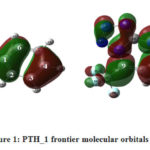 |
Figure 1: PTH_1 frontier molecular orbitals |
Observation of the frontier molecular orbitals of PTH_1 shows that
The atoms C1, C2, C3, C6, N23 and C9 being enclosed in the larger lobes of the HOMO are the atoms most susceptible to electrophilic attacks and those, contained in the larger lobes of the LUMO, to namely C9 and C7 are the sites of nucleophilic attacks.
The analysis of the frontier molecular orbitals of PTH_2 is given in Figure 2 below
 |
Figure 2: Frontier molecular orbitals of PTH_2 |
Observation of the frontier molecular orbitals of PTH_2 shows that
The atoms C1, C2, C3, C4, C5, C6 and N23 being enclosed in the larger lobes of HOMO are the atoms most susceptible to electrophilic attacks and those contained in the larger lobes of LUMO, namely C8, C7 and N16 are the sites of nucleophilic attacks.
The analysis of the frontier molecular orbitals of PTH_3 is given in Figure 3 below
 |
Figure 3: Frontier molecular orbitals of PTH_3. |
Observation of the frontier molecular orbitals of PTH_3 shows that
The atoms C1, C2, C3, C4, C5, C6 and Cl25 being enclosed in the larger lobes of HOMO are the atoms most susceptible to electrophilic attacks and those contained in the larger lobes of LUMO, namely C8, C7, N16, N21 and N23 are the sites of nucleophilic attacks.
The analysis of the frontier molecular orbitals of PTH_4 is given in Figure 4 below
 |
Figure 4: Frontier molecular orbitals of PTH_4. |
Observation of the frontier orbital map of PTH_4 shows that
The atoms C1, C2, C3, C4, C5, C6 and Br25 being enclosed in the larger lobes of HOMO are the atoms most susceptible to electrophilic attacks and those contained in the larger lobes of LUMO, namely C7 and C9 are the sites of nucleophilic attacks. The analysis of the frontier molecular orbitals of PTH_5 is given
 |
Figure 5: Frontier molecular orbitals of PTH_5. |
Observation of the frontier orbital map of PTH_5 shows that
The atoms C1, C3, C4, C5 and C6 being enclosed in the larger lobes of the HOMO are the atoms most likely to undergo electrophilic attacks and those, contained in the larger lobes of the LUMO, namely C7 , C8 and C9 are the sites of nucleophilic attacks.
The analysis of the frontier molecular orbitals of PTH_6 is given by the following figure 6
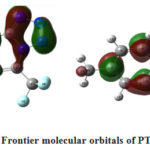 |
Figure 6: Frontier molecular orbitals of PTH_6. |
Observation of the frontier orbital map of PTH_6 shows that
The atoms C1, C2, C3, C4, C5 and C6 being enclosed in the larger lobes of the HOMO are the atoms most susceptible to electrophilic attacks and those, contained in the larger lobes of the LUMO, to namely C7 and C9 are the sites of nucleophilic attacks.
The visualization of the frontier molecular orbitals indicates for a given molecule, several sites of electrophilic and nucleophilic attacks. However, to be better situated on the most probable electrophilic and nucleophilic site, the calculation of the local Fukui indices was carried out.
Calculation of Fukui indices
The calculation of the Fukui indices of PTH_1 allows us to obtain the values recorded in Table 4.
Table 4: Values of local Fukui indices, calculated at the B3LYP / 6-311G (d, p) level, according to NPA (Natural Population Analysis) for PTH_1
|
Atome |
NPA |
|
|
f + |
f – |
|
|
C8 |
0.0036 |
0.0044 |
|
C9 |
0.1503 |
0.0404 |
|
C10 |
-0.017 |
0.0038 |
|
N17 |
0.1413 |
0.0843 |
|
C18 |
0.0076 |
-0.002 |
|
F19 |
0.0264 |
0.0145 |
|
F20 |
0.0202 |
0.0129 |
|
F21 |
0.0265 |
0.0144 |
|
N22 |
0.0235 |
0.002 |
|
N23 |
0.0443 |
0.121 |
Analysis of the data in Table 4 indicates that the N23 atom has the highest nucleophilic Fukui index for the population analysis used when C9 displays the highest electrophilic Fukui index. Therefore, any electrophilic attack will preferentially be on the N23 atom while a nucleophilic attack will be on the C9 atom.
With regard to the TPH_2 molecule, the calculation of its Fukui indices leads to the values shown in Table 5.
Table 5: Values of local Fukui indices, calculated at the B3LYP / 6-311G (d, p) level, according to NPA (Natural Population Analysis) for TPH_2.
|
Atome |
NPA |
|
|
f + |
f – |
|
|
C1 |
-0.0300 |
0.1199 |
|
C2 |
0.0064 |
0.0222 |
|
C3 |
-0.004 |
0.0587 |
|
C4 |
0.0153 |
0.1027 |
|
C5 |
-0.006 |
0.0294 |
|
C6 |
0.0184 |
0.0365 |
|
C7 |
0.1001 |
-0.0010 |
|
C8 |
-0.015 |
0.0041 |
|
C9 |
0.0982 |
0.0385 |
|
C10 |
-0.1740 |
0.0033 |
|
N16 |
0.2956 |
0.0832 |
Analysis of the data in Table 5 indicates that atom C1 has the highest nucleophilic Fukui index for the population analysis used when N16 displays the highest electrophilic Fukui index. Therefore, any electrophilic attack will preferentially be on the N16 atom when a nucleophilic attack will be on the C1 atom.
The calculation of the Fukui indices of TPH_3 leads to the values shown in table 6.
Table 6: Values of local Fukui indices, calculated at the B3LYP / 6-311G (d, p) level, according to NPA (Natural Population Analysis) for TPH_3.
|
Atome |
NPA |
|
|
f + |
f – |
|
|
C10 |
-0.0170 |
0.0030 |
|
N16 |
0.1373 |
0.0748 |
|
C17 |
0.0066 |
-0.0020 |
|
F18 |
0.0253 |
0.0133 |
|
F19 |
0.0193 |
0.0112 |
|
F20 |
0.0254 |
0.0132 |
|
N21 |
0.0242 |
0.0041 |
|
N22 |
0.0439 |
0.1041 |
|
N23 |
0.0691 |
0.1022 |
|
N24 |
0.0696 |
0.0362 |
|
Cl25 |
0.0762 |
0.1792 |
Analysis of the values in Table 6 shows that the N16 atom has the highest electrophilic Fukui index while the Cl25 atom has the highest nucleophilic Fukui index. All nucleophilic attacks will preferentially go to the N16 atom and all electrophilic attacks will go to the Cl25 atom. The calculation of the Fukui indices of TPH_4 leads to the values shown in table 7.
Table 7: Values of local Fukui indices, calculated at the B3LYP / 6-311G (d, p) level, according to NPA (Natural Population Analysis) for TPH_4.
|
Atome |
NPA |
|
|
f + |
f – |
|
|
C9 |
0.1428 |
0.0339 |
|
C10 |
-0.0170 |
0.0023 |
|
N16 |
0.1365 |
0.0699 |
|
C17 |
0.0065 |
-0.0011 |
|
F18 |
0,0250 |
0.0127 |
|
F19 |
0.0192 |
0.0104 |
|
F20 |
0.0252 |
0.0126 |
|
N21 |
0.0243 |
0.0048 |
|
N22 |
0.0436 |
0.0965 |
|
N23 |
0.0687 |
0.0943 |
|
N24 |
0.0691 |
0.0352 |
|
Br25 |
0.0889 |
0.2414 |
Analysis of the data in Table 7 indicates that the Br25 atom has the highest nucleophilic Fukui index for the population analysis used when C9 displays the highest electrophilic Fukui index. Therefore, any electrophilic attack will be on the Br25 atom while a nucleophilic attack will be on the C9 atom.
The calculation of the Fukui indices of TPH_5 leads to the values shown in table 8.
Table 8: Values of local Fukui indices, calculated at the B3LYP / 6-311G (d, p) level, according to NPA (Natural Population Analysis) for TPH_5.
|
Atome |
NPA |
|
|
f + |
f – |
|
|
C1 |
-0.0380 |
0.1441 |
|
C2 |
0.0265 |
0.0003 |
|
C3 |
0.0202 |
0.0816 |
|
C4 |
0.0728 |
0.0682 |
|
C5 |
0.0091 |
0.0483 |
|
C6 |
0.0656 |
0.0251 |
|
C7 |
0.1223 |
-0.0240 |
|
C8 |
0.0084 |
0.0028 |
|
C9 |
0.1516 |
0.0325 |
Analysis of the values in this Table 8 indicates that atom C1 has the highest nucleophilic Fukui index for the population analysis used when C9 displays the highest electrophilic Fukui index. Therefore, any electrophilic attack will be on the C1 atom while a nucleophilic attack will be on the C9 atom.
Calculation of the Fukui index of TPH_6 leads to the values shown in Table 9.
Table 9: Values of local Fukui indices, calculated at the B3LYP / 6-311G (d, p) level, according to NPA (Natural Population Analysis) for HTP 6.
|
Atome |
NPA |
|
|
f + |
f – |
|
|
C1 |
-0.0330 |
0.1296 |
|
C2 |
0.0362 |
0.0213 |
|
C3 |
0.0154 |
0.0506 |
|
C4 |
0.0836 |
0.1417 |
|
C5 |
0.0097 |
0.0193 |
|
C6 |
0.0538 |
0.0358 |
|
C7 |
0.1248 |
-0.0090 |
|
C8 |
0.0040 |
0.0033 |
|
C9 |
0.1489 |
0.0357 |
Analysis of the data in Table 9 indicates that the highest nucleophilic Fukui index value is obtained with the C4 atom from which this atom is found to be the preferential center of all electrophilic attacks while the highest electrophilic Fukui index is obtained with the C9 atom. However, the latter will be the most likely site for nucleophilic attacks.
Conclusion
At the end of our work, we retain that with the level B3LYP / 6-311G (d, p), we proceeded to the characterization of the sites of reactivity of the six (06) molecules of HTP. The values of the local Fukui indices indicated that the sites of reactivity vary depending on the substituent. Molecules with a halogen substituent have revealed that their nucleophilic attack sites are these halogens in question except fluorine whose nucleophilic site is on the C1 atom. Regarding electrophilic sites, the C9 atom is most likely for the molecules of HTP 1, HTP 4, HTP 5 and HTP 6.
The exploitation of the results obtained with the calculation of the global descriptors indicated to us that the HTP 1 molecule is the most stable of all, while the HTP 5 molecule is the most reactive with methoxy as a substituent. The substitution of halogens on the core of the HTP, indicates to us that the stability of our molecules increases with the halogens. Therefore, the more electronegative the halogen, the more stable the HTP molecules. The HTP 1 molecule has the greatest stability with hydrogen (H) as a substituent.
The calculation of the thermodynamic parameters allowed us to know that the formation of our hybrid tetrazole pyrimidine (HTP) molecules occurred spontaneously with the release of heat and reduction of disorder.
In perspective, we can consider to see what influence of temperature and pressure on the stability and reactivity of our series of molecules.
Acknowledgement
We are grateful to Professor ZIAO Nahossé, Director of the Laboratory of Thermodynamics and Physical Chemistry of the Environment, to Dr. BAMBA Kafoumba of the University NANGUI ABROGOUA (Ivory Coast), to Professor NIAMIEN Paulin Marius of the Laboratory of Constitution and Reaction of Matter of the University Félix Houphouët Boigny (Ivory Coast) for their support and motivation.
References
- Brown, G. D., Denning, D. W., Gow, N. A., Levitz, S. M., Netea, M. G., & White, T. C. (2012). Hidden killers: human fungal infections. SciTransl Med 4: 165rv13.
CrossRef - Nam, S. I; Aghebati-Maleki A., Morovati, H.; Aghebati-Maleki, L. Biomed.Pharmacother. 2019, 110, 857-868
CrossRef - Revie, N. M.; Iyer, K. R. ; Robbins, N.; Cowen, L. E. Curr. Opin. Microbiol. 2018, 45, 70-76
CrossRef - Joules, J.A.; MILLS. Blackwell Publishing House. 2000, 507-511
- Butler R. N.; Katritzky, C.W. Rees, Eds., Pergamon Press: Oxford. 1984, 5, 791
- Frederic, R B. Chem. Rev. 1947,41, 1-61
- Ostrovskii, V. A.; Koren, A. O. Heteroclycles, 2000, 53,1421
CrossRef - Amigoni, S. ;Fensterbank H.; Gaucher H. Ed Belin. 2004
- Lee, C.; Yang, W.; Parr, R. Physical Review Journals. 1988, 37, 785
CrossRef - Axel, B. D. Journal of Chemical Physics. 1993, 98, 5648
CrossRef - Bédé, A. L.; Assoma, A. B.; Yapo, K. D.; Koné, M. G.-R.; Koné, S.; Koné, M.; N’Guessan, B.; Bamba, E.-H. S. Computational Chemistry. 2018, 6, 57-70
CrossRef - Frisch, M. J.; Trucks, G. W.; Schlegel, H. B.; Scuseria, G. E.; Gaussian, Inc Gaussian 09, Revision A.02, Wallingford CT, 2009
- Kapp, J.; Remko, M.; Schleyer, P. v. R. Journal of the American Chemical Society. 1996, 118, 5745-5757
CrossRef - Johnson, B. G.; Gill, P. M.; Pople, J. A. The Journal of Chemical Physics, 1993, 98, 5612-5626.
CrossRef - Parr, R. G.; Yang, W. Annual Review Physical Chemistry. 1995, 46, 701-728
CrossRef - Coulibaly, W. K.; N’dri, J.; Koné M. G.-R., Dago, C. D.; Ambeu, C. N.; Bazureau, J.-P.; Ziao N. Computational Molecular Bioscience. 2019, 9, 49-62
CrossRef - Bohoussou, K. V.; Bénié, A.; Koné G.-R. M.; Diki, N. Y. S.; Kouassi, K. A. R.; Ziao, N.; Modern Chemistry, 2019, 7, 38-44.
CrossRef - Koopmans, T.; Physica, 1934, 104-113
CrossRef - Chattaraj, P. K.; Nath, S.; Sannigrahi, A. B.; J. Phys. Chem. 1994, 98, 9143
CrossRef - Yang, W.; Mortier, W. J.; J. Am. Chem. Soc. 1986, 108, 5708.
CrossRef

This work is licensed under a Creative Commons Attribution 4.0 International License.
About The Author
![]()
A New Edition of Web of Science
Journal Impact Factor
2022: 0.5
Five Year: 0.8
Journal is Indexed in

Cabells Whitelist
![]()


