Development and Validation of Estimation of Genotoxic Impurity (Triethyl orthoformate content) in 5-methyl-4-isoxazole carboxylic acid (5-MIA) by using GC Technique
Mohan bhatale1,2 , Neelakandan kaliyaperumal2, Gopalakrishnan Mannathusamy1 and Gurunathan ramalingam1
, Neelakandan kaliyaperumal2, Gopalakrishnan Mannathusamy1 and Gurunathan ramalingam1
1Departmentof Chemistry, Annamalai University,Annamalai Nagar, Chidambaram,India
2Emcure Pharmaceuticals Limited, Hinjawadi, Pune,11057,India
Corresponding Author E-mail: drgr_dde@rediffmail.com
DOI : http://dx.doi.org/10.13005/ojc/370212
Article Received on : 22-Mar-2021
Article Accepted on : 25-Apr-2021
Article Published : 13 Apr 2021
A simple, selective, precise and accurate Gas chromatographic method for determination of Triethyl orthoformate content (Genotoxic impurity) in 5-MIA is reported. The GC method development and validation as per the International Council for Harmonisation (ICH) guidelines Q2(R1). The effective chromatographic separations were achieved on DB-624, 60 m × 0.53 mm ID, with film thickness of 3.0 μm (Fused silica capillary column), Capillary injector temperature of 150°C, and Nitrogen Carrier gas. This method is unique as there is no UV response; hence GC Method was developed for Triethyl orthoformate. The elution was accomplished with the flow rate of 5.0 mL/min and Split Flow of 10 mL/minute. Detection was performed with FID detector (temp. 260°C) and with column oven temperature program. Methods range from limit of quantitation (LOQ) to 150% level with respect to specification concentration limit of impurity is linear and correlation coefficient of impurity is > 0.99. The linearity of Triethyl orthoformate covered from LOQ to 113 ppm (ie. LOQ to 150% of specification limit) and LOQ to 19 ppm wrt standard concentration. The limit of detection (LOD)values were observed were 2.5 ppm and limit of quantitation (LOQ) were 7.7 ppm, respectively. The parameters selected for the method validated were from international conference on harmonization guidelines, Indian pharmacopeia, USP. The percentage recovery from LOQ, 50% ,100% to 150% level of content were 87.70%, 98.60%, 102.25 and 96.59% respectively. The %RSD values were for LOQ to 150% were from 1.64%, 0.89%, 1.78 % and 1.49%. The range was covered from LOQ to 150% of standard concentration. The results of validation parameters were found in the acceptance range. Standard and sample were stable up to 30 h at when stored at room temperature. Also it was quite robust for the small change in method parameter like, change in column oven temperature
(± 5 degree). Hence from the above parameter it was concluded that the GC method with FID detector is selective, precise, linear, and robust for simultaneous estimation of Triethyl orthoformate in Drug Substances.
Triethyl orthoformate; GC-FID; Quantitative analysis; Genotoxic Impurity
Download this article as:| Copy the following to cite this article: Bhatale M, Kaliyaperumal N, Mannathusamy G, Ramalingam G. Development and Validation of Estimation of Genotoxic Impurity (Triethyl orthoformate content) in 5-methyl-4-isoxazole carboxylic acid (5-MIA) by using GC Technique. Orient J Chem 2021;37(2). |
| Copy the following to cite this URL: Bhatale M, Kaliyaperumal N, Mannathusamy G, Ramalingam G. Development and Validation of Estimation of Genotoxic Impurity (Triethyl orthoformate content) in 5-methyl-4-isoxazole carboxylic acid (5-MIA) by using GC Technique. Orient J Chem 2021;37(2). Available from: https://bit.ly/3dflGF2 |
Introduction
Leflunomide (see Fig. 1) is an immune-suppressive1 disease-modifying anti-rheumatic drug (DMARD) and, mainly use for active moderate-to-severe rheumatoid arthritis2 and psoriatic arthritis The chemical name of Leflunomide is a 4-isoxazole carboxamide, 5-methyl-N-[4-(trifluromethyl)-phenyl]. It acts inhibitor of synthesis of pyrimidine which works by preventing dihydroorotate dehydrogenase. The molecular formula and molecular weight of Leflunomide is C12H9F3N2O2 and 270.21 respectively.
Also Leflunomide3 manufactured from starting material 4-(trifluoromethyl) aniline( TFMA) and 5-Methylisoxazole-4-carboxylic acid (5-MIA). In the synthesis of the starting material 5-MIA the potential genotoxic agent Triethyl orthoformate is used. Hence, occurrence of this impurity investigated in 5-methyl-4-isoxazole carboxylic acid (5-MIA ) of Drug substance (Leflunomide).
Moto of study was aimed to design, develop, sensitive, cost effective and fully validated GC method with FID detection method, for analysis of Triethyl orthoformate impurity in Drug substances.
 |
Figure 1: Chemical structureof Leflunomide. |
Materials and Method
The sample of Leflunomide and its impurity are received from Emcure ARCH, Pune. Analytical grade sodium hydroxide, cyclohexane, sodium sulfate and purified water.Carrier Gas as a Nitrogen. Analytical balance is used as metler Toledo and using chromeleon software for data analysis and calculations. Instruments were calibrated during analytical study.
Sodium hydroxide solution
10% solution of sodium hydroxide in purified water.
Blank Solution
Add 3 mL of sodium hydroxide solution and 3 mL Cyclohexane to separating funnel. Shake well for about 1 min and allow the layers to settle down. Discard lower layer, after discarding initial 0.5 mL from upper layer, transfer about 2 mL of the upper Cyclohexane layer to 20 mL headspace vial. Add 100 mg of sodium sulfate to the vial. Shake well, wait for the solids to settle down and decant the supernatant liquid in auto sampler vial. Crimp the cap. Use this solution for analysis.
Standard and test preparation
Prepare standard solution of 12.5 ppm and sample solution with 166666 ppm using cyclohexane as diluting agent.
Pipette 5 mL of each solution in different 50 mL flash contining 20 mL cyclohexane. Dilute up to mark with cyclohexane. Pitette 3 mL of this solution and containg 3 mL sodium hydroxide solution in seperationg funnel. Shake for 1 min allow to setter layers. Discard lower layer from upper layer transfer 2 mL cyclohexane layer in 20 mL headspace vial.Add 100 mg sodium sulfate to vial.shake well to settle solids and decant supernatant liquid in auto sampler vial, inject this solution.
Method Development
As tthere is no UV response for triethyl orthoformate, hence GC Method has developed for Triethyl orthoformate.
Gas chromatography method parameters optimization
In order to achieve good resolution, better sensitivity, symmetric peak4 shape for selected drugs several trails were conducted to optimize the chromatographic method parameters (ie. Change in analytical column). The separation and analysis were done on DB-624, 60 m × 0.53 mm ID, with film thickness 3.0 μm Fused silica capillary column having with Capillary injector (with 150°C) and Nitrogen as Carrier gas.. The column temperature was set at form 50 ±2°C to 240±5°C with gradient program. Also detector temperature kept as 260°C.
The maximum response of Triethyl orthoformate was detected & was chosen for analysis. By use of above described conditions, the retention times for Triethyl orthoformate was observed as 12.5 min
(Fig. 1). Total run time of analysis was 28 minute.
The concentration limit in ppm of genotoxic impurity (Triethyl orthoformate) in drug substance derived from the TTC (Threshold of Toxicological Concern) can be calculated based on the expected daily dose to the patient using equation:
Concentration limit (ppm) = TTC [μg/day]/dose (g/day] = 1.5/0.02 = 75 ppm
The appropriate program, flow rate, gradient column oven temperature, injector temperature, detector temperature is selected by performing different trial runs of standard preparation. Method development in chromatographic conditions as follows in Table 1.
Table 1: Chromatographic conditions of method for the Triethyl orthoformate content.
|
Instrument |
GC-Perkin Elmer Turbomatrix40 with auto sampler, Perkin Elmer (Clarus 680 or equivalent) |
|
Column |
:DB-624, 60mLength × 0.53 mm ID, 3.0 µm film thickness Fused silica capillary column |
|
CarrierGas |
Nitrogen |
|
Injector temperature |
150 °C |
|
Detector |
Flame ionization detector (FID) |
|
Volume ofinjection |
1.0 μL. |
|
Detectortemperature |
260°C |
|
Attenuation |
-6(1) |
|
Detectorrange |
1 |
|
Flow rate |
1 5.0 mL-min |
|
splitflow |
10mL-min |
Moreover, gradient column oven program has been used to perform the GC analysis as, initially preparationthe temp 50°C has been used and hold for 2 min, which is then gradually changed to 240°C with ramp of 10°C min-1 and hold for 7 minute.
Results and Descusion
The ICH guideline Q2 (R1)5-8 utilized for analytical method validation study.The ICH guideline M7 (R1)9 utilized for specification limit finalization based on dose and duration.The analytical method validation parameters are described as follows.
Specificity
Slectivity study was performed to verify the absence of any interference by the components diluent. For this parameter, prepared solutions of diluent Blank, working standard solution of (12.5 μg/mL Triethyl orthoformate) and sample solution (166666 μg/mL-Triethyl orthoformate). The chromatograms are analysed at same program mentioned in method.
The represented chromatograms are shown in following Fig. 2. The chromatogram confirmed the selectivity of the method, because there were no any peaks at the retention time of selected drugs in the chromatogram of blank. The peak of Triethyl orthoformate is well separated form any other peak due to blank if any. The retention time (min) of selected impurity (Triethyl orthoformate ) in chromatograms of standard and sample solution were almost similar.
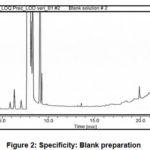 |
Figure 2: Specificity: Blank preparationthe. Click here to View figure |
 |
figure 3: Specificity: Standard preparation Click here to View figure |
 |
Figure 4: Specificity: Unspiked test Preparation Click here to View figure |
 |
Figure 5: Specificity: Spiked test Preparation |
Table 2: Specificity data
|
Impurity Name |
Individual solution |
Spiked test preparation |
|
Retention Time(minute) |
Retention Time(minute) |
|
|
Triethyl orthoformate |
Not detected |
12.51 |
Determination of limit of detection ( LOD) and limit of quantitation (LOQ)
Limit of detection (LOD) is the lowermost concentration of interested analyte in a sample solution that can be only detected but not nessasory to quantitate. In case of quantitation (LOQ), the lowest amount of interested analyte in a sample solution which can be analysed quantitatively with required precision,accuracy. In this study, LOD and LOQ concentration established by injecting various concentration levels (between 10 to 100%) of standard solutions (12.5 ppm) limit level concentrations and determined by s/n ratio. The predicated and observed LOD and LOQ data shown in Table 3.
Linearity and Range
Linearity of the method was analysed and evaluated by using known amount standard solutions of Triethyl orthofomat in Leflunomide with level of LOQ to 150% level (including 50, 80, 100 and 120%) of specification limit concentration. The conc. of LOD and LOQ, correlation coefficient, slope and intercept of the linearity results are reported in Table 4 and respective linearity graphs are represented in Fig. 6. The peak area verses concentration of interested analyte was treated by least squares linear regression analysis in which correlation coefficienst for impurity > 0.999. The %Y intercept of calibration curve not more than 5%.
Table 3: LOD, LOQ concentration and s/n ratio value of LOQ level of Triethylorthoformate
|
Impurity |
LOQ (ppm) w.r.t test |
S/N ratio LOQ level |
LOD (ppm)w.r.t test |
|
Triethylorthoformate |
7.70 |
19 |
2.54 |
Table 4: Linearity results (concentration, Averagearea , correlation coefficient, slopeand intercept of Triethyl orthoformate).
|
Linearity Levels |
Concentrati on w.r.t. (standard solution) in% |
Conc. in (ppm) |
Conc. in (ppmw.r.tSample conc.) |
AverageArea ratio of Triethylorthofor mate (n = 3) |
|
Linearity Level-1 (LOQ) |
LOQ |
1.2591 |
7.5545 |
10569.3333 |
|
Linearity Level-2 |
50 |
6.2959 |
37.7753 |
60314.0000 |
Note: (n = 3) Linearity levels LOQ to 150 % injected in triplicate
 |
Figure 6: Linearity graph of Triethyl orthoformate Precision Click here to View figure |
Precision
The precision study further sub-divided into repeatibily, intermediate precision(IP) and reproducibility(MP). In repeatibity, system precision is studied by analysing six replicates of known Triethyl orthoformate standard solution (%RSD 3.40%). The repeatability(REP) of method has been performed by injecting and analysing six individual test sample solution by spiking specification level concentration of Triethyl orthoformate. The impurity content and %RSD of the spiked sample solution was calculated.
For intermediate precision(IP) analytical activity performed on different days using different instruments, column and analysts in same laboratory.Reslult was calculated in %RSD in repeatability (n=6) along with intermediate precision (n=6). The %RSD of six spiked test preparation in repeatability for impurity (Triethyl orthofromate) is 1.81 and overall %RSD of this impurity for cumulative samples (i.e. six from repeatability and six from intermediate precision) is 3.42 respectively. The reprodubility shows collaboration study commonly applied for the standardization of methodology. The comparative data for % impurity is shown in Table 5.
Table 5: Comparative results of repeatability (REP) and intermediate precision (IP) in spiked test (75 ppm spiked Triethyl orthoformate imp )
|
Tests |
Content of Triethyl orthoformate (ppm) |
||
|
REP |
IP |
||
|
Spiked test-1 |
80.1818 |
76.8072 |
|
|
Spiked test-2 |
81.5012 |
74.1474 |
|
|
Spiked test-3 |
77.9766 |
75.9368 |
|
|
Spiked test-4 |
77.7346 |
74.1557 |
|
|
Spiked test-5 |
78.7694 |
75.2987 |
|
|
Spiked test-6 |
79.7471 |
73.2761 |
|
|
Mean (n=6) |
79.3184 |
74.9369 |
|
|
SD (n=6) |
1.4355 |
1.3130 |
|
|
RSD in%(n=6) |
1.81 |
1.75 |
|
|
Mean (n=12) |
77.1277 |
||
|
Standard Deviation (n=12) |
2.6374 |
||
|
RSD in%(n=12) |
3.42 |
||
Accuracy
The accuracy of an analytical method is the closeness of the experimental value of the substance and actual added amount of the substance in the sample matrix. Accuracy study of known impurity was carried out on triplicate sample, in the level LOQ, 50, 100 and 150% of the specification limit concentration of impurity (Triethyl orthoformate). The observed %accuracy for impurity (Triethyl orthoformate) well within acceptance criteria. The average %accuracy and its results given in Table 6.
Table 6: % Accuracy data of Triethylorthoformate impurity.
|
Impurity |
Unspikelevel |
LOQ level |
50% level |
100%level |
150%level |
|
Test sol 1 |
0.0 |
87.8723 |
98.5204 |
103.7652 |
98.2506 |
|
Test sol 2 |
0.0 |
89.0369 |
99.5136 |
100.2306 |
95.7375 |
|
Test sol 3 |
0.0 |
86.1820 |
97.7587 |
102.7650 |
95.7721 |
|
Mean |
0.0 |
87.70 |
98.60 |
102.25 |
96.59 |
|
SD |
– |
1.4354 |
0.8799 |
1.8219 |
1.4410 |
|
%RSD |
– |
.64 |
0.89 |
1.78 |
1.49 |
Solution Stability
The stability of Analytical test solution was analysed and evaluated at the room temp. on the hourly basis up to 30 hours. The %cumulative RSD of impurity (Triethyl orthoformate) was calculated for the study period of test solution. Cumulative %RSD of peak area of impurity standard solution is within acceptance criteria. This indicates that the test solution and standard solution are stable up to 30 h, when stored at room temperature.
Robustness
In robustness study, purposely altering method parameters such as change in the column oven temperature, change in flow also using the different lot of column. Considering all extreme possible variation in flow rate as well as column oven temperature. it is decided that, robustness study performed by changed by flow rate ±10% of its actual flow rate given in method. The actual flow rate of mobile phase is 5.0 mL-min, it is altered as 4.5 mL-min and 5.5 mL-min. The column oven temp changed with ±5°C from 50°C in the original analytical method it altered as 55°C and 45°C. The intermediate precision data performed on different lot number of column and shows no variation in the results. In evaluation it is observed that, the retention times are varied by ±0.2 minutes compared to actual retention times (minute). In all changed chromatographic conditions (flow rate, initial column oven temperature and different lot number of column), no significant change are observed, for the system suitability criteria and %RSD results. The values of these criteria are well within acceptable limits. The overall result of n= 8 test preparation (6 of repeatability and 2 of robustness) are given in Table 7.
Table 7: Robustness study in spiked test preparations of Triethyl orthoformate (n=8)*
|
System suitability parameters |
Triethyl Orthoformatein |
||||
|
Change in flow |
Change in Column oven temperature |
||||
|
|
5.5 mL/min |
4.5 mL/min |
55°C |
45°C |
|
|
– |
78.8451 78.2391 |
78.5077 |
78.9732 |
78.2391 |
|
|
% RSD |
1.98 |
2.46 |
1.75 |
3.00 |
|
*6 spiked test from repeatability study and 2 from robusrness study taken for comparison
Conclusion
A highly precise and accurate stability indicating GC method for the Triethyl orthoformate (Genotoxic impurity) analysis of Leflumomide an API is developed, evaluated and successfully validated with reference guideline ICH Q2(R1). Specificity shows that, Triethyl orthoformate peak is completely resolved from unknown impurities. This method is linear from LOQ to 150% level with respect to specification conc. and observed correlation coefficient of Triethyl orthoformate being greater than 0.999. In Robustness study no any significant change in the system suitability criteria ie. tailing factor and %RSD. The values of these criteria are well within acceptable limits. The Validated method shows satisfactory data for all the analysed method parameters. The present method is specific, linear, precise, selective, robust, as well as stable.
Acknowledgement
The Mohan Bhatale expresses gratitude to Dr. Mukund Gurjar, Emcure ARCH, Pune for their valuable support, inspiration and permitting this Analytical work to communication for publication.
Conflict of interest
None Conflict of interest declared.
References
- Leflunomide monograph for Professionals Drugs.com, American Society of Health-system Pharmacists.
- Leflunomide in monotherapy of rheumatoid arthrities : meta-analysis of randomized trials, Dominik Golicki., 2012, 122(1-2), 22-32.
CrossRef - Rx list, Leflunomide description., 2007.
- United States Pharmacopoeia USP USP 43–NF 38, US Pharmacopoeial Convention. General chapter<621> Chromatography General chapter <1010> Analytical Data-Interpretation and treatment.
- ICH guidelines; Q8 (R2), Harmonised Tripartite Guideline, on Pharmaceutical development. Proceedings of the International Conference on Harmonization., 2009.
- ICH guidelines; Q3A (R2), Impurities in new drug substances, in: International Conference on Harmonization, Geneva., 2006.
- ICH guidelines, Q1A(R2); Stability Testing of New Drug Substances and Products, International Council for Harmonization Geneva. (http://wwwichorg/products/guidelines/quality/article/quality-guidelines/html)., 2003.
- ICH guidelines, Q2 (R1): Validation of Analytical Procedures: Text and Methodology, International Conference on Harmonization Geneva. (http://wwwichorg/products/guidelines/quality/article/quality-guidelines/html)., 2005.
- ICH guideline M7(R1) on assessment and control of DNA reactive (mutagenic) impurities in pharmaceuticals to limit potential carcinogenic risk.

This work is licensed under a Creative Commons Attribution 4.0 International License.
About The Author
![]()
A New Edition of Web of Science
Journal Impact Factor
2022: 0.5
Five Year: 0.8
Journal is Indexed in

Cabells Whitelist
![]()


