Computational investigation of methamphetamine and its halogenated derivatives as the main source of pain and skin itching
J. Shakibayi Far
Department of Chemistry, college of Basic Sciences, Tehran science and Research Branch, Islamic Azad University, Tehran, Iran
DOI : http://dx.doi.org/10.13005/ojc/300432
Article Received on :
Article Accepted on :
Article Published : 19 Dec 2014
The structures and stability of methamphetamine and its halogenated derivatives as the main causes of increasing blood pressure, heart rate and arrhythmia chronic has been investigated Via NMR, Frequencies Calculation and CHELPG analysis. The structures of methamphetamine and halogenated derivatives were designed primarily using of Chem. Bio Draw and their geometry has been optimized at the MP2/6-31 G computational level. Frequencies calculations have been carried out to confirm that the structures obtained correspond to energetic minima. According to the obtained thermodynamic parameters, the Gibbs free energy (ΔG) and standard enthalpies (ΔH) of all these molecules are positive and they have unstable structure.Moreover the Natural Bond Orbital (NBO) analysis has provided the detailed insight into the type of hybridization and the nature of bonding in these molecules.
KEYWORDS:Methamphetamine; NMR; CHELPG; Computational
Download this article as:| Copy the following to cite this article: Far J. S. Computational investigation of methamphetamine and its halogenated derivatives as the main source of pain and skin itching. Orient J Chem 2014;30(4). |
| Copy the following to cite this URL: Far J. S. Computational investigation of methamphetamine and its halogenated derivatives as the main source of pain and skin itching. Available from: http://www.orientjchem.org/?p=6066 |
Introduction
Methamphetamineis a variant of amphetamine which has been synthesized in Japan in 1893 by Nagayoshi Nagai from the chemical ephedrine precursor [1-3]. Methamphetamine has one more methyl group in comparison to amphetamine;it is a stimulant and addictive substance that is a stronger central nervous system stimulant than amphetamines [4]. At the room temperature, methamphetamine is a clear and colorless liquid with an odor characteristic of geranium leaves and it has a chiral center with two enantiomers; In contrast, the amphetamine hydrochloride salt is odorless with a bitter taste[5-9].
Methamphetamine causes increasing in blood pressure, heart rate and arrhythmiachronic which cause weight loss, tooth decay, hallucinations, pain, and skin itching.Methamphetamine effects on the body depending on the dosage and duration of its use. In low doses, it can cause an elevated mood and increases the heart rate, blood pressure and body temperature, as well as dilate the pupil. On the other side, in high doses, it can results in heart problems, liver or kidney failure, seizures and even coma [10,11]. Moreover the animal studies show that increasing the concentration of injected methamphetamine decreased dopamine release from the striatum which leads to auto-phagocytosis of the neuritis and apoptosis of neurons in the striatum [12-19].
In recent decades, an increase in methamphetamine use has been associated with increased disability and death. So understanding the exact structure of methamphetamine and its derivate has attracted more attention in order to find the exact mechanism of methamphetamine acting in the body [20-24].
In this work, the structure of methamphetamine and halogenated derivatives has been investigated Via NMR, MP2/6-31G, NBO Calculations and CHELPG analysis and their HOMO and LUMO energies have been calculated in order to find the reactivity of these compounds. Moreover the formation energies and Gibs free energies of these compounds calculated via Gaussian software [25, 26].
Computational Methods
The structures of methamphetamine and halogenated derivatives were designed primarily using of Chem. Bio Draw 12.0 (Figure 1). The geometry of the systems has been optimized at the MP2/6-31 G of computational level [27-29]. Frequencies calculations have been carried out to confirm that the structures obtained correspond to energetic minima. All the calculations of all systems are done in the gas phase by density functional theory (DFT) using MP2 method and the standard 6-31G basis set using Gaussian 09(Gaussian 09, Revision A.02).The total energy, electronic and nuclear energies were calculated for the three studied compounds, as well as HOMO, LUMO energies and the difference between then (∆E). Finally obtained results were compared to other derivatives.Mullikan charges on the atoms, dipole moments, and geometry parameters such as bond lengths were also determined by the same method. The energy barrier for rotation around the tow bond in the methamphetamine molecule is also assessed using Gaussian software [30-36].
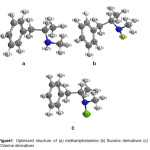 |
Figure1: Optimized structure of (a) methamphetamine (b) fluorine derivatives (c) Chlorine derivatives Click here to View figure |
Results and Discussion
Geometries
The structures of methamphetamine and halogenated derivatives were designed using of Chem. Bio Draw 12.0 and the obtained results were compared to other derivatives(Figure 1). The optimization, NBO, NMR and CHELPG calculations of the whole systems are done by density functional theory (DFT) using MP2 method and the standard 6-31 G basis set using Gaussian09 [37,38].
The optimized geometrical parameters, such as Dipole moment (Debye), nitrogen charge, structure formation energy (HF: kcal/mole), enthalpies (ΔH) and Gibbs free energy (ΔG) are listed in Table 1.As shown in Table 1, the chlorine derivate has the lowestΔG is 99.7307677 Kcal/mol and the methamphetamine molecule has the largest ΔG is 121.29689 Kcal/mol.The molecule has the lowestΔH is 148.321687 Kcal/mol and the methamphetamine molecule has the largest ΔH is 153.048689 Kcal/mol.Therefore, the formation of chlorinated derivatives requires less energy [39, 40].
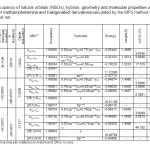 |
Table1: Occupancy of natural orbitals (NBOs), hybrids, geometry and molecular properties and CHELPG of methamphetamine and halogenated derivatives calculated by the MP2 method with 6-31G(d) basis set. Click here to View table |
Rotation energy barrier
The energy barrier for rotation around the tow bond in the methamphetamine molecule is also assessed using Gaussian software and the results are represented in Table 2 and Figure 2.
As shown in Table 2 rotation -61.3182 degree around the N10-C11 bond has the lowest energy state is the most stable for methamphetamine [41, 42].
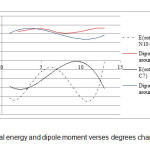 |
Figure2: Potential energy and dipole moment verses degrees changing Click here to View figure |
Table2. The calculated energy barrier and dipole moment of rotationaround bonds N10-C11 and C6-C7.
|
Rot1 (C8-N10-C11-H26) |
Rot2 (C5-C6-C7-C8) |
||||
| Angstroms and Degrees | E(k(Cal/mol) | Dipole moment | Angstroms and Degrees | E(k(Cal/mol) | Dipole moment |
|
-76.3181 |
-1.93521 |
1.5846 |
-96.2876 |
-1.3394 |
1.5217 |
|
-61.3182 |
-2.28435 |
1.6619 |
-81.2875 |
-1.62046 |
1.7067 |
|
-46.3181 |
-2.00311 |
1.7511 |
-66.2877 |
-1.66513 |
1.8218 |
|
-31.3181 |
-1.21214 |
1.8158 |
-51.2877 |
-1.49703 |
1.8153 |
|
-16.3184 |
-0.35071 |
1.7985 |
-36.2877 |
-1.17443 |
1.7328 |
|
-1.3184 |
0 |
1.6636 |
-21.2876 |
-0.75589 |
1.617 |
|
13.6819 |
-0.36922 |
1.5272 |
-6.2877 |
-0.32937 |
1.4923 |
|
28.682 |
-1.13101 |
1.5213 |
8.7123 |
-0.07455 |
1.3835 |
|
43.682 |
-1.90245 |
1.5765 |
23.7123 |
0 |
1.2813 |
|
58.682 |
-2.27939 |
1.6655 |
38.7124 |
-0.10837 |
1.2157 |
|
73.6819 |
-2.05387 |
1.7711 |
53.7124 |
-0.49905 |
1.2172 |
|
88.6818 |
-1.29089 |
1.8653 |
68.7124 |
-0.96265 |
1.303 |
|
103.6817 |
-0.3839 |
1.7596 |
83.7124 |
-1.36337 |
1.3993 |
Energies
Various energies such as the equilibrium energy, HOMO-LUMO band gap corresponding to methamphetamine and halogenated derivatives after the geometry optimization are summarized in Table 1. The HOMO and LUMO energies are so important since the basic electronic parameters related to the energy level of frontier orbitals in a molecule and their resulting energy gap. Also both HOMO and LUMO are the main orbital take part in chemical stability [43-46].
As shown in Table1,the molecule has the largest energy gap is -132.8053 Kcal/mol and the methamphetamine molecule has the lowest energy gap is -34.7199Kcal/mol.Therefore, fluorinated derivative is less reactive in comparison to methamphetamine because the role of a nucleophile to share electrons[47-49].
NMR
In this section, the NMR shielding tensors, isotropic shielding (σiso) and anisotropic shielding(σaniso), σ11, σ22, σ33 of methamphetamine and its halogenated derivate has been investigated using MP2/6-31G level in the gas phase. Hamiltonian Chemical shielding is given by following equation (Mehring,1983) [50]: H = yhσBσI Where γ is magneto-gyric ratio, B0 is magnetic field and I is nuclear spin operator.The σ in this equation called NMR chemical shielding tensor whose elements describe the size of chemical shielding as a function of molecular orientation with respect to the external magnetic field.
Quantum chemical calculations yield chemical shielding tensors in the principal axis system with the order of σ33 >σ22 >σ11 hence, Eqs. (1) And (2) are used to convert the calculated chemical shielding tensors to the absolute isotropic ( ) and anisotropic ( ) chemical shielding parameters:
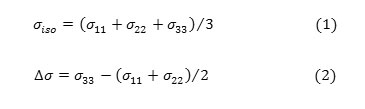
The shielding tensor asymmetry parameter (η) is given by equation 3.
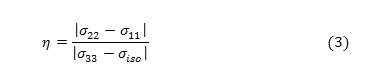
The span ( ) describes the maximum width of the powder pattern and the skew ( )describes the amount and orientation of the asymmetry of the tensor [43, 50-52].
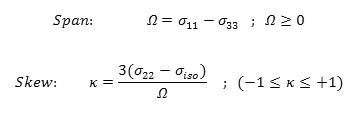
Table 3 presents the computed chemical shift for selected hydrogen, nitrogen and carbon atoms. According to this table, isotropic shielding value (σiso ) for N atom of molecule is a negative value, because F is electronegative. N of molecule has the lowest isotropic shielding and the highest anisotropic shielding value, respectively, because Cl is electronegative and large atom.
NBO Analysis
Population analysis is a mathematical way of partitioning a wave function or electron density into charges on the nuclei, bond orders, and other related information.A full NBO analysis is obtained in Gaussian when using the POP=NBO keyword.
Natural bond analysis provides an efficient way to study intra and intermolecular bonding and interaction among bonds, and also provides a convenient basis for investigating charge transfer or conjugative interactions in molecular systems.
NBOs are an orthonormal set of localized “maximum occupancy” orbitals whose leading N/2 members (or N members in the open-shell case) give the most accurate possible Lewis-like description of the total N-electron density(Theoretical Chemistry Institute of the University of Wisconsin).
Table 1 lists the calculated occupancies of natural orbitals for methamphetamine and its halogenated derivate as well as calculated Natural bond hybrids.
For each electron donor (i) and acceptor (j), the stabilization energy E(2) associated with the delocalization i→jis estimated as
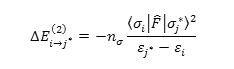
Where is the effective orbital Hamiltonian and < σj |F|σj* >2 is the Fock matrix of element i and j NBO orbitals, εj* and εj are the energies of i and j* NBOs j* and is the population of the donor orbital .
Table 4 lists the selected values of second order perturbation theory analysis of Fock matrix at MP2/6-31G level of theory as well as the selected values of the calculated second order interaction energy (E2) between donor–acceptor orbitals in methamphetamine and halogenated derivatives.According to these results, the strongest interactions are the electron donations from a π c-c to the anti bonding acceptor π* c-c orbitals.According to calculations, the nitrogen atom forms three single bond (sigma bond) with two carbon atoms C8 and C11 ( σcε – N10 and σN10 – C11 ) and a hydrogen atom H23 (σH23 -N10) a lon pair orbital (Lp), and a CR orbital. As seen from Table 3, the LP hybrid is an sp5.87 for the molecule,Sp2.52hybrid for the molecule, and Sp2.85For the molecule.
 |
Table3: Computed chemical shifts for selected atoms Click here to View table |
 |
Table4:Second-order interaction energy (E2, kcal/mol) between donor and acceptor orbitals in methamphetamine and halogenated derivatives Click here to View table |
Charges from Electrostatic Potentials Using a Grid (ChelpG)
In this method, the molecular electrostatic potential is observable and is used for ChelpG population analysis method.Though the molecular electrostatic potential is observable (can be measured) in the population analysis, it is computed from the wave function of the system. The most notable disadvantage of ChelpG is that it does not work well for large systems, especially those with “inner” atoms since the molecular electrostatic potential is a surface characteristic (Ramachandran et al., 2008).53 The results of ChelpG are collected in Table 1. As it can be seen in this table, of these molecules has the lowest ∆V chelpg is -2.7023j/c in the H-Mta , -1.79231 j/c in the F-Mta, and -2.39221 j/v in the Cl-Mta.
Conclusion
In the present work, the theoretical analyses of methamphetamine and its halogenated derivate have been performed and the Dipole moment (µ) and energy of structure formation (HF)of these molecules have been obtained. According to these results has lowest and methamphetamine has highest dipole moment and HF.
Also according to the obtained thermodynamic parameters, it has been found that the amount of Gibbs free energy (ΔG) and standard enthalpies (ΔH) of methamphetamine and its halogenated derivate are positive value, therefore methamphetamine and its halogenated derivate are unstable structure. Also the chemical shifts have been calculated using quantum mechanics.
Moreover the Natural Bond Orbital (NBO) analysis has provided the detailed insight into the type of hybridization and the nature of bonding in methamphetamine and its halogenated derivate. Finally the Charges from Electrostatic Potentials Were also calculated and the results compared to different derivatives.
References
- Nagai, T.; Kamiyama, S. International Journal of Legal Medicine. 1988, 101(3), 151-159.
- Lineberry, T.W.Mayo Clinic Proceedings. 2006, 81(1), 77-84.
- Meredith,C.W.; Jaffe, C.;Ang-Lee, K.; Saxon A.J. Harvard Review of Psychiatry. 2005, 13(3),141-154.
- Molitor,F.;Truax, S.; Ruiz, J. West J Med.1998, 168, 93–97.
- Chang, L.;Alicata, D.;Ernst. T. Addiction. 2007,102(Supple 1), 16–32.
- Mollaamin,F.; Monajjemi,M.Physics and Chemistry of Liquids. 2012, 50(5), 596–604
- Monajjemi, M.; Rajaeian,E.; Mollamin,F.; Naderi, F.; Saki,S. Physics and Chemistry of Liquids. 2008, 46, 299-306.
- Mollaamin,F.; Varmaghani,Z.;Monajjemi, M.Physics and Chemistry of Liquids. 2011, 49(3), 318–336
- Monajjemi,M.; Honarparvar, B. H.;Haeri, H.;Heshmat, M. Russian Journal of Physical Chemistry C.2006, 80(1), S40-S44.
- Westfall, D.P.; Westfall, T.C.Miscellaneous Sympathomimetic Agonists.In Brunton LL, Chabner BA, Knollmann BC. Goodman & Gilman’s Pharmacological Basis of Therapeutics (12th ed.). New York: McGraw-Hill. 2010. ISBN 978-0-07-162442-8.
- Chomchai, C.; Na Manorom, N.; Watanarungsan.P.;Yossuck P.; Chomchai S. Southeast Asian J. Trop. Med. Public Health. 2010, 35 (1): 228–231. PMID 15272773.
- Ricaurte, G.A.; Schuster, C.R.; Seiden, L.S.Brain Research. 1980, 193, 153–163.
- Wagner, G.C.; Ricaurte, G.A.; Johanson, C.E.; Schuster, C.R.; Seiden, L.S. Neurology. 1980, 30, 547–550.
- Villemagne, V.; Yuan, J.; Wong, D.F.; Dannals, R.F.; Hatzidimitriou, G.; Mathews, W.B.; Ravert, H.T.;Musachio, J.; McCann, U.D.; Ricaurte, G.A. The JournalofNeuroscience.1998, 18, 419–427.
- Volkow, N.D., Chang, L., Wang, G.J., Fowler, J.S., Franceschi, D., Sedler, M., Gatley, S.J., Miller, E., Hitzemann, R., Ding, Y.S.The Journal of Neuroscience 2001, 21, 9414.
- Monajjemi, M.; Mollamin,F.Journal of Computational and Theoretical Nanoscience, 2012, 9(12) 2208-2214 .
- Mollamin,F.; Monajjemi, M.; MehrzadJ. Fullerenes, Nanotubes, and Carbon Nanostructures. 2014, 22: 738-751,
- Monajjemi, M.; Robert Wayne, Jrand James E. Boggs, Chemical Physics. 2014, 433, 1-11.
- Ghalandari, B.; Monajjemi, M.; Mollaamin, F.Journal of Computational and Theoretical Nanoscience. 2011, 8: 1212-1219.
- Monajjemi, M.;FarahaniN.; Mollaamin,F. Physics and Chemistry of Liquids. 2012, 50(2), 161-172.
- Mollaamin, F.; Najafpour, J.;Ghadami, S.;Ilkhani, A. R.;Akrami, M. S.;Monajjemi, M. J. Comput. Theor .Nanosci. 2014, 11, 1290-1298
- Monajjemi, M.; Mollaamin, F.;Karimkeshteh, T. J. Mex. Chem. Soc. 2005, 49(4), 336-340.
- ahanA.;Monajjemi, M. ActaBiotheor. 2011, 59, 291–312.
- Monajjemi, M.; Mollaamin,F. J Clust Sci. 2012, 23, 259–272
- Monajjemi, M.; Khaleghian, M. J Clust Sci. 2011, 22, 673–692
- Monajjemi, M.; Faham R.;Mollaamin,F. Fullerenes, Nanotubes, and Carbon Nanostructures. 2012, 20, 163-169.
- Møller,C.; Plesset, M. S.Phys. Rev.1934, 46, 0618-22
- Frisch, M. J.; A.Pople, J. S.; Binkley, J.J. Chem. Phys. 1984, 80, 3265.
- Frisch, M. J. et al. Gaussian 09, Revision A.02, 2009. Gaussian, Inc., Wallingford CT, 2009. Gaussian, Inc., Wallingford CT.
- Monajjemi,M.; JafariAzan M.; Mollaamin F. Fullerenes, Nanotubes, and Carbon Nanostructures. 2013, 21(6), 503–515.
- Yahyaei,H.;Monajjemi, M.;Aghaie,H., Zare K.Journal of Computational and Theoretical Nanoscience. 2013, 10(10), 2332-2341.
- Monajjemi, M.; Boggs, J.E. J. Phys. Chem A. 2013, 117, 1670.
- Ardalan, T.;Ardalan,P.;Monajjemi, M. Fullerenes, Nanotubes, and Carbon Nanostructures. 2014, 22, 687–708,
- Monajjemi, M.Chemical Physics. 2013, 425, 29-45.
- Yahyaei,h.;Monajjemi, M.Fullerenes, Nanotubes, and Carbon Nanostructures. 2014, 22, 346-361.
- Monajjemi, M.; Lee, V. S.; Khaleghian, M.; Honarparvar, B.; Mollaamin,F. J. Phys. Chem. C. 2010, 114, 15315-15330.
- Monajjemi, M. Karachi, N.;Mollaamin, F.Fullerenes, Nanotubes, and Carbon Nanostructures. 2014, 22, 643-662.
- Monajjemi, M.Struct Chem. 2012, 23, 551-580.
- Monajjemi, M.; Heshmat, M.; Haeri,H. H.Biochemistry (Moscow). 2006, 71, S113-S122.
- Monajjemi, M.;Sobhanmanesh, A.; Mollaamin, F. Fullerenes, Nanotubes, and Carbon Nanostructures. 2013, 21, 47-63.
- Monajjemi, M.;Baheri, H.; Mollaamin, F.Journal of Structural Chemistry. 2011, 52(1), 54-59.
- Monajjemi, M.;SeyedHosseini, M.; Mollaamin, F.Fullerenes, Nanotubes, and Carbon Nanostructures. 2013, 21, 381-393.
- Gunase karan, S.; Balaji, R. A.; Kumaresan, S. Can. J. Anal. Sci. Spectrosc. 2008, 53, 149.
- Monajjemi, M.;Yamola, H.; Mollaamin, F. Fullerenes, Nanotubes, and Carbon Nanostructures. 2014, 22, 595–603.
- Mollaamin,F.;Monajjemi, M. Journal of Computational and Theoretical Nanoscience. 2012, 9(4), 597-601.
- Monajjemi, M.; Chegini,H.; Mollaamin,F.; Farahani, P.;Fullerenes, Nanotubes, and Carbon Nanostructures. 2011, 19, 469-482.
- Monajjemi, M.; Falahati,M.; Mollaamin, F. Ionics. 2013, 19, 155-164.
- Mollaamin, F.;Shahani pour, K.;Ilkhani.; A. R.;Sheckari, Z.; Monajjemi, M. Russian Chemical Bulletin, International Edition. 2012, 61(12), 2193-2198.
- Monajjemi, M.; Afsharnezhad,S.; Jaafari, M.R.; Abdolahi, T.; Nikosade, A.;Monajjemi, M. Russian Journal of physical chemistry A.2007, 2,1956-1963.
- Mehring, M.Principles of High Resolution NMR in Solids. Springer, Berlin.1983.
- Monajjemi, M.; Razavian, M.H.; Mollaamin, F.; Naderi, F.; Honarparvar, B.Russian Journal of Physical Chemistry A.2008, 82 (13), 2277-2285.
- Defalque, R.J.; Wright,A.J.Bull. Anesth. Hist. 2011, 29(2): 21–24, 32.
- Ramachandran, K.I.;DeepaGopakumar, Krishnan Namboori. Springer Berlin Heidelberg. 2008, 253-255.

This work is licensed under a Creative Commons Attribution 4.0 International License.
![]()
A New Edition of Web of Science
Journal Impact Factor
2022: 0.5
Five Year: 0.8
Journal is Indexed in

Cabells Whitelist
![]()


