Structural and stability investigation of the anticancer drug Cyclophosphamide via quantum chemical calculations :A nanotube drug delivery
Z. Felegari
Department Of Chemistry, Science and Research Branch, Islamic Azad University, Tehran, Iran
DOI : http://dx.doi.org/10.13005/ojc/300447
Article Received on :
Article Accepted on :
Article Published : 18 Dec 2014
Cyclophosphamide is a medicine used to interfere with the growth and spread of tumor cells and treat cancers and autoimmune disorders.This work reports the study of anticancer drugs with density functional theory (DFT) and electronic structures.Its structure was optimized with B3LYP/6-311G* level in the gas phase and different solvents (SCRF calculation). NBO analysis,NMR parameter,thermodynamic properties,HOMO and LUMO,HOMO-LUMO band gap, and the electronic chemical potential (µ) were calculated. The results indicated that the Cyclophosphamide in water solvent is more stable than the gas phase orother solvents.
KEYWORDS:Cyclophosphamide; NBO; NMR parameter; HOMO-LUMO gap; DFT
Download this article as:| Copy the following to cite this article: Felegari Z. Structural and stability investigation of the anticancer drug Cyclophosphamide via quantum chemical calculations :A nanotube drug delivery. Orient J Chem 2014;30(4). |
| Copy the following to cite this URL: Felegari Z. Structural and stability investigation of the anticancer drug Cyclophosphamide via quantum chemical calculations :A nanotube drug delivery. Available from: http://www.orientjchem.org/?p=5990 |
Introduction
Cyclophosphamide(Procytox or Cytoxan) is a drug used in the treatment of cancer, andis in a class of drugs known as alkylating agents. Cyclophosphamide (C7H15Cl2N2O2P ) is also used to treat bronchogenic carcinoma, small cell lung carcinoma, and other types of cancer [1-3].
It is obvious ,drug delivery technology modifies drug release profile, absorption, distribution and elimination for the benefit of improving product efficacy and safety, as well as patient convenience and compliance[4-7].
For this reason many protein and peptide drugs have to be delivered by injection or a nanoneedlearray.Today efforts in the area of drug delivery include the development of targeted delivery in which the drug is only active in the target area of the body [8-11].
Cyclophosphamide is biotransformed principally into the liver to active alkylating metabolites by a mixed function of microsomal oxidase system. These metabolites interfere with the growth of susceptible rapidly proliferatingmalignant cells. The mechanism of action is thought to involve the cross-linking of tumor cell DNAs. Cyclophosphamide is well absorbed after oral administration, with a bioavailability of greater than 75%. The unchanged drug has an elimination half-life of 3-12 hours. It is eliminated primarily in the form of metabolites,but 5-25% of the dose is excreted in urine in its original form. Several cytotoxic and noncytotoxic metabolites have been detected in urine and plasma. Concentrations of metabolites was maximized in plasma in 2-3 hours after an intravenous dose. Plasma protein binding of unchanged drug is low, but some metabolites are bound to an extent of greater than 60%. It has not been demonstrated that any single metaboliteis responsible for either the therapeutic or toxic effects of cyclophosphamide. Although elevated levels of metabolites of cyclophosphamide have been observed in patients with renal failure, increased clinical toxicity in such patients has not been demonstrated [12-17].
An alkylating agent adds an alkyl group (CnH2n+1) to DNA molecules that link sit with this method,while DNA replication is inhibited.DNA is one of the most important biological molecules targeted by many smaller molecules (proteins representing extremely important targets as well). Many of scientists have focused on biological applications of inorganic systems so nano sensors based on biology in biomedical devices and bioreactors have considerable applied in the last years [18-20].
Also,during the past decades, molecules binding with DNAA have been seriously taken into account[21,22]. A lot of investigations of the interaction of drug molecules withDNA have been studied[23-27].
The integration of biological processes and synthesize molecules with fabricated structures presented also both electronic control and bio-electronically driven nano-assembly [28-30].
As a specific example, hollow cylinders that made of many sheets of carbon atoms to mean carbon nano tubes have recommended for use in nervous systems as prosthetic implants, and obtaining this goal requires the incorporation of fully functioning nano-electronic and biological systems [31,32].
In this work letter, we report our study on the stability of the anticancer drug Cyclophosphamide in the gas phase and different solvents. We found that the Cyclophosphamide be have differently in the gas and solvent phase.
Computational method
We modeled the structure of Cyclophosphamide with Gauss view 5.0[33], and then optimized it in the gas phase and different solvents, such as Water, DMSO, Ethanol, and Methanol.
All calculations were carried out with the Gaussian 09 program[34]. The calculations of systems containing C,H, N, P, O and Cl are explained by the standard 6-311 G (d) basis set function of the Density Functional Theory (DFT)[35,36].
After optimization, we calculated the NMR parameters and NBO. The population analysis has been performed by the natural bond orbital method at B3LYP/6-311 G (d) level of theory using natural bond orbital (NBO)[37].
[38,39].NBO analysis used the B3LYP method and 6-311G* basis set, and the output is obtained for molecule. Finally, the highest occupied molecular orbital (HOMO), lowest unoccupied molecular orbital (LUMO), energy gaps, and thermodynamic properties have been discussed[40-43]
We calculated the NMR parameters at the levels of B3LYP/6-311G* theory[44,45], and theoretically explored the effects of solvent (water, DMSO, methanol, ethanol) on the structure of Cyclophosphamide. All the relative energy values and NMR shielding parameters were calculated by assuming that the gauge includes the atomic orbital (GIAO) method.The Gauge Including Atomic Orbital (GIAO)[46-48]approach was used[49,50].The abinitio GIAO calculations of NMR chemical shielding tensors were performed using the DFT method [51].The chemical shielding tensors were calculated by the GAUSSIAN 09 program[52].
Results and Discussion
In our study, we performed quantum calculations on the structure of Cyclophosphamide, which is an important anticancer drug. Therefore, HF and DFT methods, with 6-311 G*basis set, were employed to investigate the structures, optimization, and energy minimization of Cyclophosphamide (Fig.1) in the gas phase and different solvents (Water, DMSO, Ethanol and Methanol) that have been summarized in Tables 1 a and 1 b.We take this medication to the Quantum computation phase solvents,such as water, DMSO, ethanol and methanol were used in the following ways to deduce the effect of solvents on the drug.Also, we effectively investigated the solvent on this drug, and optimized it at the B3LYP levels of theory, with 6-311G* basis set being summarized in Table 1b. According to the values listed in Table 1b, it indicates that the solvent effect the bond lengths, so (C2-C3), (C3-H8), (P12-N13) and (P12-N25) in water is shorter than the gas phase and other solvents.Also, (P12-O14) in water is longer than the gas phase and other solvents, which proves that electron-donor atoms decreases bond lengths, while the electron-pull atoms increases it.
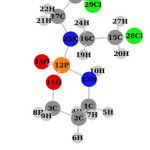 |
Fig1: Structure of Cyclophosphamide Click here to View figure |
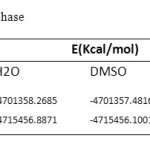 |
Table1a: Optimizes energy for each phase Click here to View table |
Table 1b.Bond Lengths (A0) for Cyclophosphamide in Gas phase and different solvent
| Atom | Gas | Water | DMSO | Ethanol | Methanol |
| C2-C3 | 1.52413 | 1.52067 | 1.52077 | 1.52085 | 1.52078 |
| C3-H8 | 1.09499 | 1.09421 | 1.09423 | 1.09424 | 1.09423 |
| P12-O14 | 1.48259 | 1.49236 | 1.49220 | 1.49179 | 1.49197 |
| P12-N13 | 1.69209 | 1.67533 | 1.67558 | 1.67628 | 1.67596 |
| P12-N25 | 1.66929 | 1.66929 | 1.66935 | 1.66937 | 1.66934 |
The highest occupied molecular orbital (HOMO), the lowest unoccupied molecular orbital (LUMO), and the energy gap was calculated by B3LYP/6-311G*method, and are provided in Table 2. The HOMO represents the ability to donate an electron,while LUMO acts as an electron acceptor, representing the ability to obtain an electron. The electron transition absorption corresponds to the transition from the ground to the first excited state, and is mainly described by electron excitation from HOMO to LUMO.
The energy gap between HOMO and LUMO is a critical parameter in determining molecular electrical transport properties[53,54].The HOMO and LUMO of Cyclophosphamide are represented in Table 2 and Fig.2. The energy gap and electron potential in water solvent are larger than other solvents.A large gap implies high stability, while a small gap implies low stability. The high stability in turn indicates low chemical reactivity, and a small gap indicates high chemical reactivity. Therefore, the results confirm the stability of Cyclophoshamide in water.
Table2. Obtained some Parameter by B3LYP/6-311G* Level
| Parameter | Gas | Water | DMSO | Ethanol | Methanol |
| EHOMO(eV) | -0.25396 | -0.25495 | -0.25455 | -0.25447 | -0.25450 |
| ELUMO(eV) | 0.00542 | 0.00435 | 0.00439 | 0.00447 | 0.00443 |
| Energy gap(eV) | 7.0581 | 7.0559 | 7.0460 | 6.2827 | 7.0458 |
| µ(eV) | -3.3815 | -3.4095 | -3.4036 | -3.0197 | -3.4024 |
 |
Fig2: The HOMO and LUMO of Cyclophosphamide in Gas phase and different solvent Click here to View figure |
The isotropic chemical shielding (σiso) and anisotropy shielding (Δσ) for O14, Cl28, and Cl29 of Cyclophosphamide calculated in the gas phase and different solvents (Fig.1) are summarized in Table 3, as the highest and the lowest density of charges is concentratedonthese atoms (Fig.3,4).
 |
Fig3: Electron density from Total SCF Density (isoval=0. 0004) Click here to View figure |
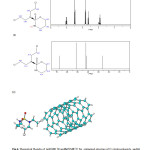 |
Fig4: Theoretical Results of (a)NMR 1H and(b)NMR13C for optimized structure of Cyclophosphamide and(c) SWCNT (5,5) armchair – Cyclophosphamide complex . Click here to View figure |
Table 3. NMR parameter’s value σiso (ppm) and Δσ (ppm) of O14, Cl28, Cl29 of Cyclophosphamide in Gas phase and different solvent at the level of B3LYP/ 6-311G* basis set at the DFT theory
| B3LYP/6-311G* | Isotropic | Anisotropy | B3LYP/ | Isotropic | Anisotropy |
| (σISO) | (Δσ) | 6-311G* | (σISO) | (Δσ) | |
| O14 (Gas) | 175.6586 | 103.2645 | Cl28 (DMSO) | 864.0924 | 55.085 |
| P12 (Gas) | 291.2206 | 266.0028 | Cl29 (DMSO) | 868.8859 | 40.5847 |
| Cl28 (Gas) | 855.7205 | 396.3267 | O14 (Ethanol) | 183.1043 | 24.9665 |
| Cl29 (Gas) | 857.2639 | 397.8573 | P12 (Ethanol) | 287.5821 | 54.858 |
| O14 (Water) | 183.368 | 39.9917 | Cl28 (Ethanol) | 863.8796 | 260.1108 |
| P12 (Water) | 287.3822 | 26.4779 | Cl29 (Ethanol) | 868.4794 | 260.1108 |
| Cl28 (Water) | 864.1848 | 55.1154 | O14 (Methanol) | 183.1961 | 40.3907 |
| Cl29 (Water) | 869.0759 | 258.4659 | P12 (Methanol) | 287.5194 | 25.4539 |
| O14 (DMSO) | 183.2886 | 40.8805 | Cl28 (Methanol) | 863.9808 | 54.9611 |
| P12 (DMSO) | 287.4358 | 25.9317 | Cl29 (Methanol) | 868.6892 | 259.5927 |
The blue regions show the most electron deficient regions, while the red color areas show the most electron accumulation regions.Therefore, the O14, Cl28, and Cl 29is regarded as important. The chemical shift value of O14 in water s
We calculated the thermodynamic functions, such as constant volume molar heat capacity (CV), enthalpy (H), Gibbs free energy (G), total energy (E), and entropy (S) for Cyclophosphamide in the gas phase and different solvents obtained from the the oretical method by B3LYP/6-311G* and its respective values listed in Table4. All of the thermodynamic data supply helpful information for a study on the Cyclophosphamide. They can be used to compute other thermodynamic energies according to the relationships of thermodynamic functions[55].
We compared the gas phase and solvent effects on the thermodynamic parameter of Cyclophosphamide. Table 8 showed that the total energy (Etotal), entropy (S), and constant volume molar heat capacity (CV) values are positive,while energy (Etotal), enthalpy (H) and G Gibbs’ free energy are negative values.These calculations were repeated in various solvents with different dielectric constants.The results showed that the stability of Cyclophosphamide is reduced by the decreasing polarisability of the solvents.The highest stability is observed for water, with ε=78.39, while the lowest is for Ethanol, with ε=24.55.
The natural bond orbital analysis provides the accurate possible natural Lewis structure. The results of the interaction is a loss of occupancy from the concentration of electron NBO of the idealized Lewis structure in an empty non-Lewis orbital. A careful examination of all possible interactions between ‘‘filled’’ (donor) Lewis-type NBOs and‘‘empty’’ (acceptor) non-Lewis NBOs allows us to estimate their energetic importance via the second-order perturbation theory. For each donor(i) and acceptor (j), the stabilization energy E (2)is associated with the delocalization[56]. The strengths of these delocalization interactions, E (2), are estimated by the second order perturbation theory.Some of the significant donor–acceptor interactions and their second order perturbation stabilization energies E (2) of Cyclophosphamide is given in Table 4. This section shows some of the donor–acceptor interactions and their second order perturbation energies E (2) for Cyclophosphamide[35].
Table 4.The calculated thermodynamic parameters (E to talk cal/Mol, CV kcal /Mol, S kcal/molK, H kcal/Mol, G kcal/Mol and E kcal/Mol) of Cyclophosphamide in gas phase and different solvent
| Parameter | Gas | Water | DMSO | Ethanol | Methanol |
|
ε |
− |
78.39 |
47 |
24.55 |
32.63 |
|
E total |
157.328 |
158.154
|
158.157 |
158.163 |
158.160 |
|
CV |
56.393 |
56.518 |
56.512 |
56.502 |
56.507 |
|
S |
124.291 |
130.669 |
130.586 |
130.466 |
130.534 |
|
H
|
-1797.494975
|
-1797.515549
|
-1797.515266
|
-1797.514664
|
-1797.514966
|
|
G
|
-1797.554030
|
-1797.577634
|
-1797.577312
|
-1797.576652
|
-1797.576987
|
|
E |
-1797.495920 |
-1797.516493 |
-1797.516210 |
-1797.515608 |
-1797.515910 |
The most important interaction between “filled” (donor) Lewis-type NBO and “empty” acceptor) non-Lewis is reported in Table 5, with the level of B3LYP/ 6-311G* basis set at the DFT theory. The electron density is transferred from lone pair LP (2) O14 to anti–bonding σ* (P12-N13), where the interaction is seen to provide a strong stabilization 19.59 KCal/mol. This strong stabilization denotes larger delocalization[48].Finally, we reported the Energy and Natural Hybrid Orbital (NHO) for P12-N13 bonding of Cyclophosphamide in Table 6. According to Table 6, in the P12-N13 bond, BD=0. 5276 SP2.94 d 0.8+0.8495 SP2.35 was reported. Polarization coefficients of the P12-N13 bond are P12=0. 5276 and N13=0. 8495, the size of these coefficients shows the importance of the hybrid N13 in the formation of the bond[57,58].
Table 5. Donor and acceptor NBO for Cyclophosphamide and the level ofB3LYP/ 6-311G*in different solvents
| Donor | Acceptor | E2 (kcal/Mol) | Donor | Acceptor | E2 |
| NBO (i) | NBO (j) | NBO (i) | NBO (j) | (kcal/Mol) | |
| (Gas) LP (1) O14 | BD* (1) | 1.14 | (DMSO) | BD* (1) | 24.97 |
| P12 -N 13 | LP (3) O14 | O11 – P12 | |||
| (Gas) LP (2) O14 | BD* (1) | 19.84 | (Ethanol) | BD* (1) | 1.09 |
| P12 – N13 | LP (1) O14 | P12 – N13 | |||
| (Water) LP (1) O14 | BD* (1) | 1.09 | (Ethanol) | BD* (1) | 19.75 |
| P12 – N13 | LP (2) O14 | P12 – N13 | |||
| (Water) LP (2) O14 | BD* (1) | 19.98 | (Ethanol) | BD* (1) | 25.00 |
| P12 – N13 | LP (3) O14 | O11 – P12 | |||
| (Water) LP (3) O14 | BD* (1) | 24.97 | (Methanol) | BD* (1) | 1.09 |
| O11 – P12 | LP (1) O14 | P12 – N13 | |||
| (DMSO) LP (1) O14 | BD* (1) | 1.09 | (Methanol) | BD* (1) | 19.70 |
| P12 – N13 | LP (2) O14 | P12 – N13 | |||
| (DMSO) LP (2) O14 | BD* (1) | 19.96 | (Methanol) | BD* (1) | 24.99 |
| P12 – N13 | LP (3) O14 | O11 – P12 |
E (2) means energy of hyperconjugative interactions.
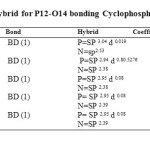 |
Table6: Energy (kcal/Mol) and hybrid for P12-O14 bonding Cyclophosphamide Click here to View table |
Conclusion
In the present work, we study the stability of Cyclophosphamide in the gas phase and different solvents. After optimization, the obtained data showed that the Cyclophosphamide is stable in water. Also, the energy gap of HOMO-LUMO confirms this stability.The σiso value of O14 in water solvent is higher than theσiso value in other solvents. This means that electron density around O14 in water solvent is higher compared to other solvents.According to NBO analysis E (2) in water, it is higher than other solvents, and the thermodynamic parameters in water are higher,which again indicates the greater stability in water.Finally, our studies in the gas phase, and different solvents showed the Cyclophosphamide in water solvent is more stable than the gas phase and other solvents.
References
- Shanafelt TD, Lin T, Geyer SM, et al., Cancer 2007, 109(11): 2291-8.
- Young SD, Whissell M, Noble JC, Cano PO, Lopez PG, Germond CJ., Clinical Cancer Research 2006, 12 (10): 3092–8.
- Nicolini A, Mancini P, Ferrari P, et al., Biomedicine & Pharmacotherapy 2004, 58 (8): 447–50.
- M. Monajjemi, J.E Boggs, J. Phys. Chem A. 2013, 117, 1670.
- H. Yahyaei& M. Monajjemi, Fullerenes, Nanotubes, and Carbon Nanostructures, 2014, 22: 346–361.
- Donelli MG, Bartosek I, Guaitani A, et al., Cancer Treatment Reports 1976, 60(4): 395–401.
- M. Monajjemi, V. S. Lee, M. Khaleghian, B. Honarparvar, F.Mollaamin, J. Phys. Chem C., 2010, 114, 15315.
- H. Yahyaei , M. Monajjemi , H. Aghaie , and K. Zare, Journal of Computational and Theoretical Nanoscience, 2013, 10(10), 2332–2341.
- M. Monajjemi, Struct Chem., 2012, 23, 551-580.
- Ramsey-Goldman R, Mientus JM, Kutzer JE,Mulvihill JJ, Medsger TA., The Journal of Rheumatology 1993, 20(7): 1152–7.
- Majid Monajjemi, Chemical Physics,2013, 425, 29-45.
- Singh G, Fries JF, Williams CA, Zatarain E, Spitz P, Bloch DA., The Journal of Rheumatology 1991, 18(2): 188–94.
- M. Monajjemi, F. Naderi, F. Mollaamin, and M. Khaleghian, J. Mex. Chem. Soc. 2012, 56(2), 207-211
- F. Mollaamin , F. Najafi , M. Khaleghian , B. Khalili Hadad & M. Monajjemi, Fullerenes, Nanotubes, and Carbon Nanostructures, 2011, 19: 653–667.
- M. Monajjemi, M. Khaleghian, J Clust Sci., 2011, 22, 673.
- Lohrmann HP., Oncology 1984, 41(3): 180-4.
- M. Monajjemi , H. Yamola& F. Mollaamin, Fullerenes, Nanotubes, and Carbon Nanostructures, 2014, 22: 595–603.
- F. Mollaamin , M. Monajjemi & J. Mehrzad , Fullerenes, Nanotubes, and Carbon Nanostructures, 2014, 22: 738–751.
- M.Monajjemi, S. Afsharnezhad, M.R. Jaafari, T. Abdolahi, A. Nikosade and H. Monajjemi. Russian Journal of physical chemistry A, 2007, 2,1956-1963.
- M. Monajjemi , A. Sobhanmanesh , & F. Mollaamin, Fullerenes, Nanotubes, and Carbon Nanostructures, 2013, 21: 47–63.
- Krishna, A. G., D. V. Kumar, et al., Biochimica et Biophysica Acta (BBA) – General Subjects 1998, 1381(1): 104-112.
- Glendening, E., J. Badenhoop, et al., “NBO 5.0; Theoretical Chemistry Institute, University of Wisconsin: Madison, WI, 2001, 2004.
- Reed, A. E., L. A. Curtiss, et al., Chemical Reviews 1988, 88(6): 899-926.
- M. Monajjemi , N. Karachi & F. Mollaamin, Fullerenes, Nanotubes, and Carbon Nanostructures, 2014, 22: 643–662.
- B. Ghalandari, M. Monajjemi, and F. Mollaamin, Journal of Computational and Theoretical Nanoscience, 2011, 8, 1212–1219.
- Majid Monajjemi, Robert Wayne, Jrand James E. Boggs, Chemical Physics, 2014, 433, 1-11.
- T. Ardalan , P. Ardalan& M. Monajjemi, Fullerenes, Nanotubes, and Carbon Nanostructures, 2014, 22: 687–708.
- M. Monajjemi, R. Faham& F. Mollaamin, Fullerenes, Nanotubes, and Carbon Nanostructures, 2012, 20: 163–169.
- M. Monajjemi ; H. Chegini ; F. Mollaamin ; P. Farahani, Fullerenes, Nanotubes, and Carbon Nanostructures, 2011, 19: 469–482.
- F. Mollaamin, J. Najafpour, S. Ghadami, A. R. Ilkhani, M. S. Akrami, and M. Monajjemi J. Comput. Theor. Nanosci. 2014, 11: 1290-1298.
- M. Monajjemi , M. SeyedHosseini& F. Molaamin, Fullerenes, Nanotubes, and Carbon Nanostructures, 2013, 21: 381–393
- M. Monajjemi & J. Najafpour, Fullerenes, Nanotubes, and Carbon Nanostructures, 2014, 22: 575–594.
- Kang, M. S., S. H. Kang, et al., Chemical Communications 2012, 48(75): 9349-9351.
- Frisch, M., G. Trucks, et al. “Gaussian 09, revision B. 01.” Gaussian, Inc., Wallingford, CT, 2010.
- Dadkhah, A., Iranian Rehabilitation Journal 2014, 12(19): 1-10.
- F. Mollaamina; Z. Varmaghanib; M. Monajjemi, Physics and Chemistry of Liquids, 2011, 49(3), 318–336
- M. Khaleghian ; M. Zahmatkesh ; F. Mollaamin; M. Monajjemi, Fullerenes, Nanotubes, and Carbon Nanostructures, 2011, 19: 251–261.
- Fatemeh Mollaamin & Majid Monajjemi, Physics and Chemistry of Liquids, 50(5): 2012, 596–604.
- Glendening, E. D., C. R. Landis, et al., “Natural bond orbital methods.” Wiley interdisciplinary reviews: computational molecular science 2012, 2(1): 1-42.
- M. Monajjemi,and M. Ahmadianarog, Journal of Computational and Theoretical Nanoscience 2014, 11(6), 1465-1471.
- Boumpas DT, Austin HA, Vaughn EM, et al. Lancet 1992, 340(8822): 741-5.
- M. Monajjemi , and F. Mollaamin , Journal of Computational and Theoretical Nanoscience, 2012, 9(12), 2208-2214.
- F. Mollaamin and M. Monajjemi ,Journal of Computational and Theoretical Nanoscience , 2012, 9(4), 597-601.
- Monajjemi, M., E. Rajaeian, et al., Physics and Chemistry of Liquids 2008, 46(3): 299-306.
- Monajjemi, M., H. Chegini, et al., Fullerenes, Nanotubes, and Carbon Nanostructures 2011, 19(5): 469-482.
- Schreckenbach, G. and T. Ziegler., The Journal of Physical Chemistry 1995, 99(2): 606-611.
- Bohmann, J. A., F. Weinhold, et al., The Journal of chemical physics 1997, 107(4): 1173-1184.
- Weinhold, F., Journal of computational chemistry 2012, 33(30): 2440-2449.
- Schreckenbach, G. and T. Ziegler., The Journal of Physical Chemistry A 1997, 101(18): 3388-3399.
- Chen, Z., C. S. Wannere, et al., Chemical reviews 2005, 105(10): 3842-3888.
- Kaupp, M., M. Bühl, et al., Calculation of NMR and EPR parameters: theory and applications, John Wiley & Sons, 2006.
- Becke, A. D., A. A. Arabi, et al., Canadian Journal of Chemistry 2010, 88(11): 1057-1062.
- M Arivazhagan& J Senthil Kumar,,Indian Journal of Pure & Applied Physics 2012, 50, 363-373.
- Bonness, S., H. Fukui, et al., Chemical Physics Letters 2010, 493(1): 195-199.
- Zeng, X.-L., X.-L. Zhang, et al. Chemosphere 2013, 91(2): 229-232.
- Glendening, E. D., C. R. Landis, et al., “Natural bond orbital methods.” Wiley interdisciplinary reviews: computational molecular science 2012, 2(1): 1-42.
- Weinhold, F. and R. A. Klein., “What is a hydrogen bond? Mutually consistent theoretical and experimental criteria for characterizing H-bonding interactions.” Molecular Physics 2012, 110(9-10): 565-579.
- Huttunen KM, Raunio H, Rautio J “Prodrugs—from serendipity to rational design”. Pharmacological Reviews 2011, 63(3): 750-71.

This work is licensed under a Creative Commons Attribution 4.0 International License.
![]()
A New Edition of Web of Science
Journal Impact Factor
2022: 0.5
Five Year: 0.8
Journal is Indexed in

Cabells Whitelist
![]()


