Synthesizing New Pyrimidinone Derivatives and Their Respective Biological Structure Assessment
Asma M. Elsharif*
College of Science, Department of Chemistry, Imam Abdulrahman Bin Faisal University, Dammam 31113, Saudi Arabia.
Corresponding Author E-mail: aelsharif@iau.edu.sa
DOI : http://dx.doi.org/10.13005/ojc/350221
Article Received on : 03-02-201
Article Accepted on : 05-04-2019
Article Published : 29 Apr 2019
Barbituric acid is converted into a pyrimidinone-incorporated pyrazolyl moiety (1), which is a key starting material. 1 can be converted into pyrimidine dione, isoxazole, pyrimidopyrimidine, and pyranopyrimidine by reacting with hydrazine hydrate and/or phenyl hydrazine, hydroxyl amine, urea, thiourea, guanidine, ethyl acetoacetate and ethyl cyanoacetate. Acylation of 1 gave an important key intermediate (7), which was condensed to form chalcone, which then underwent cycloaddition into cyclohexenes (8-13). Some newly synthesized compounds were screened as anti-diabetic agents and exhibited significant activity. These freshly manufactured compounds were characterized using different methods. These compounds showed significant activity as anti-diabetic agents, especially compound 4b, with IC50= 13.54 μg/ml, which is very close to that of the standard acarbose (IC50= 12.87 µg/ml). Additionally, these compounds showed cytotoxic inhibition activity against the colon carcinoma (HCT116), hepatocellular carcinoma (HEPG2), and breast carcinoma (MCF7) cells; compounds 11, 4b, and 10 showed the best activity, with IC50 = 19.3, 2.6, and 5 μg/ml, respectively.
KEYWORDS:Activity; Anti-Diabetic; Barbituric; Cytotoxic; Pyrazolyl; Pyrimidinone
Download this article as:| Copy the following to cite this article: Elsharif A. M. Synthesizing New Pyrimidinone Derivatives and Their Respective Biological Structure Assessment. Orient J Chem 2019;35(2). |
| Copy the following to cite this URL: Elsharif A. M. Synthesizing New Pyrimidinone Derivatives and Their Respective Biological Structure Assessment. Orient J Chem 2019;35(2). Available from: https://bit.ly/2PAprHY |
Introduction
Barbituric acid is a heterocyclic compound derived from pyrimidine. Pyrimidinones constitute a very important class of six-membered heterocyclic compounds that are widely distributed because of their importance as anti-HIV agents,1 and anti-microbial agents2,3
Barbituric acid, one of the most important naturally occurring heterocyclic compounds, serves as the fundamental compound in barbiturate drugs and is involved in a wide variety of biological activities like – Urease Inhibitors,4 adipogenesis of 3T3-L1,5 anti-diabetic and antibacterial agents,6 PPARγ activators,7 protease inhibitors,8 potential antimicrobial,9 an anesthetic drugs.10 This high activity may be due to the reactive chemical structure (pyrimidine trione). Barbituric acid is a parent compound of drugs called barbiturates.
Barbiturates have attracted considerable focus among researchers due to its involvement in different biological activities like – antioxidant,11 antibacterial agents.12 As a continuation of our previous work,13 we hope to convert barbituric acid into new heterocyclic compounds, with a focus on the application of these compounds by way of being an effective precursor in the process of preparing various new molecules of heterocyclic bio-active compounds bearing bioactive pyrazolyl,14 isoxazoles,15 pyranoes, cyclohexene16 moieties for the purpose of increasing the biological reactivity capability in the molecules. Now, the newly manufactured compounds get screened for use as anti-diabetic and anti-cancer agents.
 |
Scheme (A) |
Material and Methods
All commercial reagents; barbituric acid, ethyl acetoacetate, ethyl cyanoacetate, and hydrazine hydrate were obtained from Sigma-Aldrich. These regents were then put to use without using a purifying process. Melting temperature of the regents was then measured through use of an electro-thermal digital apparatus without making any correction. Recording of KBr (IR spectra) was done by use of the system Perkin Elmer Spectrum RXIFT-IR. Measurement of 1HNMR, 13C-NMR was done through use of Varian Gemini 850 MHZ instrument alongside TMS.
5-((5-chloro-4-methyl-1-phenyl-1H-pyrazol-3-yl)methylene)pyrimidine-2,4,6(1H,3H,5H)-trione (1)
Barbituric acid (0.01 mol) was introduced stirred to the mixture of sodium ethoxide (0.5 gram in 10 milliliter solution of ethanol) and pyrazolone (0.01 mol) at 27°C. Afterwards the whole solution was mixed for 60 minutes at the temperature of 100 degree-Celsius. Once the solution cooled down, filtration was performed of the separated solid, washed clean using water, dried down to remove moisture, and recrystallized using EtOH.
Yield is 74% and melting point (mp) 187-188°C.
Elemental Analysis of 1 C15H11ClN4O3 (330) (%) Calculated: C, 54.49; H, 3.37; Cl, 11.01; N, 17.02; Found: C, 54.43; H, 3.35; Cl, 10.98; N, 16.99.
FTIR spectra showed bands at 1625 cm-1, 1735, 1720, 1700 cm-1, 3340cm1, for √ C=N, √ C=O, √ NH respectively. Ion peak was noted at /z 330(M+, 35.5Cl) (12.5%), 332 (M+, 37.5Cl) (5.21%). The 1H-NMR exhibited signal bands δ ppm at 10.03(s,2H,2xNH), 6.85-8.04(m,5H,ArH), 5.52(s,1H,CH=CH), 2.31(s,3H,CH3), 13C-NMR: 164.96, 162.10, 152.45(C=O), 141. 68 (C=N), 135.65, 129.46, 126.54, 124.48, 123.66 C-aromatic, 129.23,126.75 (C=CH), 126.11(C-Cl), 5.16(CH3).
3-(5-chloro-4-methyl-1-phenyl-1H-pyrazol-3-yl)-2H-pyrazolo[3,4 d]pyrimidine-4,6(5H,7H)-dione (2a), 3-(5-chloro-4-methyl-1-phenyl-1H-pyrazol-3-yl)-2-phenyl-2H-pyrazolo[3,4-d]pyrimidine-4,6(5H,7H)-dione (2b)
Reflux (0.02 mol) of 1, hydrazine hydrate (0.02 mol) in ethanol (30 ml) for 6 hours. Once the solution cooled down, filtration was performed of the separated solid, washed clean using water, dried down to remove moisture, and recrystallized using EtOH. Yield 84% and the melting point (mp) 201-203°C.
Yield of 2b 65% and the melting point (mp) 273-274°C.
Elemental Analysis of 2a C15H11ClN6O2 (342) (%) Calculated: C, 53.57; H, 3.22; Cl, 10.34; N, 24.53; Found: C, 53.60; H, 3.20; Cl, 10.35; N, 24.55.
Analysis of 2b C21H15ClN6O2(418) (%)Calculated: C, 60.31; H, 3.70; Cl, 8.46; N, 20.11; Found: C, 60.11; H, 3.89; Cl, 8.48; N, 20.09.
FTIR spectra of 2a showed bands at 1602 cm-1, 1735, 1720 cm-1, 3340cm1 for √ C=N, √ C=O, √NH respectively, while for 2b; 1620 cm-1, 1725, 17210 cm-1, 1450 3340 cm-1 for √ C=N, √ C=O, √ C=C, √ NH respectively. Ion peak was noted at m/z 342(M+, 35.5Cl) (3.22%), 344 (M+, 37.5Cl) (8.45), and for 2b m/z 418(M+, 35.5Cl) (13.75%), 420(M+, 37.5Cl), (9.36%). The 1H-NMR showed signal bands δ ppm for 2a at 10.12(s,3H,3xNH), 7.09-7.92(m,5H,ArH), 2.01(s,3H,CH3), while for 2b at 10.07(s,2H,2xNH), 6.97-8.02(m,10H,ArH), 2.12(s,3H,CH3), 13C-NMR for 2a: 158.96, 155.05(C=O), 140.05, 138.55 (C=N), 134.07, 129.08(C=C), 133.25(C-N), 128.68, 125.54, 123.43, C-aromatic, 124.98(C-Cl), 9.06(CH3), for 2b: 159.06, 154.55 (C=O), 139.75, 138.05(C=N), 135.07, 132.18(C=C), 131.95(C-N), 129.55, 128.68, 127.46, 125.54, 126.43, 125.87 C-aromatic, 126.08(C-Cl), 6.76(CH3).
3-(5-chloro-4-methyl-1-phenyl-1H-pyrazol-3-yl)isoxazolo[3,4-d]pyrimidine-4,6(5H,7H)-dione (3)
Reflux (0.012 mol) of 1 and (0.012 mol) of hydroxylamine hydrochloride in pyridine (10 milliliter) for 6 hours. pour into ice/HCl. Once the solution cooled down, filtration was performed of the separated solid, washed clean using water, dried down to remove moisture, and recrystallized using EtOH. Yield is 59% and melting point (mp) 230-232°C.
Analysis of 3 C15H10ClN6O3(343) (%)Calculated: C, 52.42; H, 2.94; Cl, 10.31; N, 20.35; Found: C, 52.41; H, 2.91; Cl, 10.30; N, 20.36.
FTIR spectra of 3 showed bands at 1615 cm-1, 1725, 1690cm-1, 1210 cm-1, 3340cm-1 for √ C=N, √ C=O, √ C-O, √ NH respectively. The ion peak of the solution was noted using a mass spectrum and it was at m/z 343(M+, 35.5Cl) (22.5%), 345 (M+, 37.5Cl), (17.21%). The 1H-NM showed bands δppm at 10.13(s,2H,2xNH), 6.88-8.20(m,5H,ArH), 2.26(s,3H,CH3), 13C-NMR: 154.96, 152.45 (C=O), 149. 98 (C=N), 129.46, 126.54, 123.66 C-aromatic, 128.23, 127.75 (C=CH), 125.88(C-Cl), 6.16(CH3).
5-(5-chloro-4-methyl-1-phenyl-1H-pyrazol-3-yl)pyrimido[4,5 d]pyrimidine-2,4,7(1H,3H,4aH)-trione (4a), 5-(5-chloro-4-methyl-1-phenyl-1H-pyrazol-3-yl)-7-thioxo-4a,7-dihydropyrimido[4,5-d]pyrimidine-2,4(1H,3H)-dione (4b) ,5-(5-chloro-4-methyl-1-phenyl-1H-pyrazol-3-yl)-7-imino-4a,7-dihydropyrimido[4,5-d]pyrimidine-2,4(1H,3H)-dione (4c)
In (20 ml) glacial acetic acid reflux (0.012 mol) of 1, combined with guandine, thiourea, and urea HCl for 5 hours. Once the solution cooled down, filtration was performed of the separated solid, washed clean using water, dried down to remove moisture, and recrystallized using EtOH. Yield 74% and melting point (mp) 214-215°C.
Analysis of 4a C16H11ClN6O3(370) (%)Calculated: C, 50.83; H, 2.99; Cl, 9.56; N, 21.68; Found: C, 50.87; H, 2.97; Cl, 9.53; N, 21.67.
Analysis of 4b C16H11ClN6O2S (386) (%) Calculated: C, 50.01; H, 2.87; Cl, 10.11; N, 22.73; S, 8.29; Found: C, 49.70; H, 2.86; Cl, 10.20; N, 22.72; S, 8.27.
Analysis of 4c C16H12ClN7O2(396) (%) Calcd C, 51.97; H, 3.25; Cl, 9.57; N, 26.52; Found: C, 52.00; H, 3.25; Cl, 9.58; N, 26.49.
FTIR spectra of 4a showed bands at 1612 cm-1, 1725, 1710, 1696 cm-1, 3328 cm-1 for √ C=N, √ C=O, √ NH respectively, while for 4b at 1605 cm-1, 1725, 1721 cm1, 1410 ,3340cm-1 for √ C=N, √ C=O, √ C=S, √ NH respectively and for 4c at 1618 cm1, 1730, 1715 cm-1, 3328cm-1 for √ C=N, √ C=O, √ NH.
The ion peak of the solution was noted using a mass spectrum and it was at m/z 370(M+,35.5Cl) (35.22%), 372 (M+, 37.5Cl) (18.05) and for 4b m/z 386(M+, 35.5Cl) (33.75%), 388 (M+, 37.5Cl) and for 4c m/z 396(M+, 35.5Cl) (12.25%), 398 (M+, 37.5Cl), (7.25%) (10.25%). The 1H-NMR showed signal bands δppm for 4a at 10.09(s,2H,2xNH), 8.02(s, H,NH pyrimidine), 6.97-7.73(m,5H,ArH), 2.25(s,3H,CH3), while for 4b at 10.02 (s,3H,3xNH), 7.03-8.04(m, 5H,ArH), 2.01(s,3H,CH3) and for 4c at 10.29(s,3H,3xNH), 6.80-7.83(m, 5H,ArH), 2.10(s,3H,CH3), 13C-NMR for 4a: 158.96, 156.54,155.05 (C=O), 151.05, (C=N), 144.07, 139.08(C=C), 133.25(C-N), 128.68, 125.54, 123.43, C-aromatic, 124.98(C-Cl), 9.06(CH3), for 4b:159.06, 154.55(C=O), 139.75, 138.05(C=N), 135.07, 132.18(C=C), 129.85(C-N), 128.68, 125.54, 124.25C-aromatic, 125.88(C-Cl), 5.76(CH3) and for 4c: 160.06, 156.85(C=O), 152.75, 148.95(C=N), 162.01, 141.07, 139.18(C=C), 129.85(C-N), 128.68, 125.54, 124.25 C-aromatic, 125.88(C-Cl), 6.76(CH3).
6-acetyl-5-(5-chloro-4-methyl-1-phenyl-1H-pyrazol-3-yl)-7-hydroxy-6,7 dihydro-1H-pyrano[2,3-d]pyrimidine-2,4(3H,8aH)-dione (5)
Reflux in 20 ml ethanol, (0.012 mol) of 1, ethyl acetoacetate (0.012 mol) for 5 hours. Once the solution cooled down, filtration was performed of the separated solid, washed clean using water, dried down to remove moisture, and recrystallized using EtOH. Yield of 5 is 69% and melting point (mp) 222-223°C.
Analysis of 5 C19H17ClN4O5(416) (%) Calculated: C, 55.51; H, 3.99; Cl, 7.90; N, 13.44; Found: C, 55.56; H, 3.90; Cl, 7.98; N, 13.46.
FTIR spectra of 5 showed bands at 1210 cm-1, 1615 cm-1, 1735, 1710-1, 3340 cm-1 for √ C-O, √ C=N, √ C=O, √ NH. The ion peak of the solution was at m/z 416(M+, 35.5Cl) (11.5%), 418 (M+, 37.5Cl), (7.21%). The 1H-NMR: showed signal bands δ ppm at 10.18(s,2H,2xNH), 7.77-7.45(m,5H,ArH), 3.75(s,1H,CHCO), 2.40(s,3H, COCH3), 2.18(s,6H,2xCH3), 13C-NMR: 159.26, 153.85, 151.75(C=O), 139.98 (C=N), 128.26, 125.84, 122.76C-aromatic, 128.23, 127.75 (C=CH), 125.88(C-Cl), 6.16, 28.01(CH3).
5-(5-chloro-4-methyl-1-phenyl-1H-pyrazol-3-yl)-2,4,7-trioxo-2,3,4,6,7,8a-hexahydro-1H-pyrano[2,3-d]pyrimidine-6-carbonitrile (6)
In (20 ml) ethanol, reflux (0.012 mol) of 1 and (0.012 mol) of ethyl cyanoacetate for the duration of 5 hours. Once the solution cooled down, filtration was performed of the separated solid, washed clean using water, dried down to remove moisture, and recrystallized using EtOH. Yield of 6 is 63% and melting point (mp) 253-254°C.
Analysis of 6 C18H12ClN5O4 (397) (%) Calculated: C, 56.35; H, 2.98; Cl, 9.12; N, 17.61; Found: C, 56.40; H, 2.99; Cl, 9.20; N, 17.59.
FTIR spectra of 6 showed bands at 1615 cm-1, 1725, 1717-1, 1210 cm-1, 2234 3340cm-1 for √ C=N, √ C=O, √ C-O, √C≡N, √ NH respectively. The ion peak of the solution was noted using a mass spectrum and it was at m/z 397(M+,35.5Cl) (19.05%), 399 (M+, 37.5Cl), (2.21%). The 1H-NMR showed signal bands δppm at 10.04(s,2H,2xNH), 6.97-7.82 (m,5H,ArH), 5.01(s,1H,CHO), 3.56(s,1H,CHCN), 2.04(s,3H, CH3), 13C-NMR: 169.26, 157.25,155.45 (C=O), 139. 98 (C=N), 126.06, 123.87, 122.09 C-aromatic, 126.98(C-Cl), 115.89 (C≡N), 5.16 (CH3).
1-acetylpyrimidine-2,4,6(1H,3H,5H)-trione (7)
Stirring (0.012 mol) of compound 1, (0.012 mol) acetyl chloride in a water container for 3 hours and then pouring the whole solution in ice results in compound 7. Once the solution cooled down, filtration was performed of the separated solid, washed clean using water, dried down to remove moisture, and recrystallized using EtOH. Yield of 7 is 81% and melting point (mp) 197-198°C.
Analysis of 7 C6H6N2O4 (170) (%) Calculated: C,41.67; H, 3.58; N, 16.45; Found: C,41.40; H, 3.56; N, 16.46.
FTIR spectra of 7 showed bands at 1725, 1717, 1690 cm -1, 3340cm1 for √ C=O, √ NH. The ion peak of the solution was noted using a mass spectrum and it was m/z 170(M+) (20.05%). The 1H-NMR showed signal bands δppm at 10.22(s,1H, NH), 3.02(s,COCH2CO), 2.24(s,3H, CH3), 13C-NMR: 170.26,156.25,154.45, 152.23 (C=O), 45.07(CH2), 5.27 (CH3).
1-(3-(5-chloro-4-methyl-1-phenyl-1H-pyrazol-3-yl)acryloyl)pyrimidine-2,4,6(1H,3H,5H)-trione (8)
Stir in ice bath pyrazolone ethanolic solution (0.12 mol) and (0.04 mol) of 7 and then adding 5ml of NAOH in a 10% aqueous concentration in a drop-system for 30 minutes. Once the solution cooled down, filtration was performed of the separated solid, washed clean using water, dried down to remove moisture, and recrystallized using EtOH. Yield of 8 is 71% and melting point (mp) 229-230°C.
Analysis of 8 C17H13ClN4O4 (372) (%) Calculated: C, 54.87; H, 3.54; Cl, 10.05; N, 14.99; Found: C, 54.89; H, 3.55; Cl, 10.12; N, 14.87.
FTIR spectra of 8 showed bands at 1602 cm-1, 1645 cm-1,1690, 1725cm-1, 3340cm-1 for √ C=N, √ C=C, √ C=O, √ NH respectively. The ion peak of the solution was noted using a mass spectrum and it was m/z 372(M+, 35.5Cl) (22.05%), 374 (M+, 37.5Cl) (5.71%). The 1H-NMR showed signal bands δppm at 10.16(s,1H, NH), 7.02-8.01 (m, 5H,ArH), 5.56-5.48(d,2H,CH=CH), 3.01(s,2H,CH2CO), 2.02(s,3H, CH3), 13C-NMR: 169.26, 157.25, 155.45, 151.65 (C=O), 139. 98 (C=N), 130.25, 126.06, 123.87, 122.09, 118.23 C-aromatic, 134.24, 125.56(C=C alkene) 125.44(C-Cl), 4.86 (CH3).
6-ethyl 6-(5-chloro-4-methyl-1-phenyl-1H-pyrazol-3-yl)-2-oxo-4-(2,4,6-trioxotetrahydropyrimidin-1(2H)-yl)cyclohex-3-enecarboxylate (9)
Reflux (0.01 mol) of compound 8 in 20 milliliter of ethanol, 0.5 sodium metal in ten milliliter of ethanol (sodium ethoxide) and ethyl acetoacetate for six hours. Afterwards, the solution is kept overnight to cool down and upon recrystallization, the ethanol is separated from the solid content. Yield of 9 is 82% and melting point (mp) 255-256°C.
Analysis of 9 C23H21ClN4O6 (484) (%) Calculated: C, 56.01; H, 3.99; Cl, 6.97; N, 11.55; Found: C, 56.32; H, 3.74; Cl, 6.95; N, 11.56.
FTIR spectra of 9 showed bands at 1190 cm-1, 1602 cm-1, 1690,1753 cm-1, 3340 cm-1 for √ C-O, √ C=N, √ C=O, √ NH. The ion peak of the solution was noted using a mass spectrum and it was m/z 484(M+, 35.5Cl) (18.64%), 486 (M+, 37.5Cl) (11.02%). The 1H-NMR showed signal bands δppm at 10.04(s,1H, NH), 7.14-8.12 (m, 5H,ArH), 3.57(q,2H,COCH2CH3), 3.44(q,1H,CHCHCH2), 3.28(s,2H, CH2CO), 3.22(d,1H, CHCHCH2), 2.62(d,2H, CHCHCH2), 2.07(s,3H, CH3), 1.55(t,3H, COCH2CH3). 13C-NMR 172.05, 169.8, 158.25, 156.45, 153.65 (C=O), 141.02 (CN), 129.25, 124.95, 123.87, 122.09, 117.23 C-aromatic, 136.45, 126.89(C=C alkene) 124.44(C-Cl), 61.54, 41.89, 34.27(CH2)16.85, 5.24 (CH3).
1-(5-(5-chloro-4-methyl-1-phenyl-1H-pyrazol-3-yl)-3-oxo-4-(5-thioxo-4,5-dihydro-1H-1,2,4-triazol-3-yl) cyclohex-1-en-1-yl) pyrimidine-2,4,6(1H,3H,5H)-trione (10)
Reflux (0.012 mol) of 9 and (0.012 mol) of thiosemicarbazide, in 20 milli liter of ethanol, for six hours. After that, the solid content in crystalized form is separated from the ethanol solution. Yield of 10 is 67% and melting point (mp) 285-286°C.
Analysis of 10 C22H16ClN7O4 S (511) (%) Calculated C, 51.61; H, 3.56; Cl, 6.97; N, 19.15, S, 6.30; Found: C, 51.64; H, 3.57; Cl, 6.92; N, 19.13, S, 6.28.
FTIR spectra of 10 showed bands at 1410 cm-1 ,1608 cm-1, 1690, 1715 cm1, 3270 cm-1 for √ C=S, √ C=N, √ C=O, √ NH respectively. The ion peak of the solution was noted using a mass spectrum and it was m/z 511(M+, 35.5Cl) (11.75%), 513 (M+, 37.5Cl) (2.01%). The 1H-NMR showed signal bands δ ppm at 10.20(s,1H,NH), 7.98(s,2H,2xNH), 7.03-7.97 (m, 5H,ArH), 4.23(s,1H,CH=C), 3.56(q,1H,CHCHCH2), 3.34(d, 1H, COCHCH), 3.08 (s,2H, COCH2CO), 2.12(s,2H, CHCH2CN), 1.95(s,3H, CH3). 13C-NMR (DMSO-d6): 182.34(C=S) 175.25, 165.25, 155.27, 151.45 (C=O), 144.34, 141.23 (CN), 127.67, 125.05, 123.87, 122.09C-aromatic, 136.15, 124.55(C=C alkene), 125.44(C-Cl), 68.75 (CH2), 5.24 (CH3).
1-(5-(5-chloro-4-methyl-1-phenyl-1H-pyrazol-3-yl)-4-(5-hydrazinyl-4H-1,2,4-triazol-3-yl)-3-oxocyclohex-1-en-1-yl)pyrimidine-2,4,6(1H,3H,5H)-trione (11)
Reflux (0.01 mol) of 10, and (0.012 mol) hydrazine hydrate compound (0.012 mol) in 20 milliliter of ethyl alcohol for six hours. After that, the solid content in crystalized form is separated from the cooled down ethanol solution. Yield of 11 is 65% and melting point (mp) 255-256°C.
Analysis of 11 C22H20ClN9O4 (509) (%) Calculated: C, 51.84; H, 4.01; Cl, 6.95; N, 24.72; Found: C, 51.85; H, 4.12; Cl, 6.94; N, 24.74.
FTIR spectra of 11 showed bands at 1618 cm-1, 1715, 1730cm-1, 3330, 3340cm1 for √ C=N, √ C=O, √ NH2 respectively. The ion peak was determined using a mass spectrum and it was m/z 509(M+, 35.5Cl) (22.75%), 511 (M+, 37.5Cl) (9.58%). The 1H-NMR(DMSO-d6) displayed signal bands δ ppm at 10.28(s,1H, NH), 8.79(s,1H,NH triazole), 6.98-7.87(m, 5H,ArH), 4.89(t,1H,NHNH2), 4.09(s,1H,CH=C), 3.82(d, 1H, COCHCH), 3.51(q,1H, CHCHCH2), 3.22(d,2H, CH2CH), 3.08(s,2H,COCH2CO), 2.55(d,2H, NHNH2), 2.32(s,2H, CH2N), 2.13(s,3H, CH3). 13C-NMR (DMSO-d6): 185.25,165.25, 156.45,153.65 (C=O), 148.62,141.02 (CN), 127.67, 125.05,123.87, 122.09C-aromatic, 139.05, 122.44(C=C alkene), 125.44(C-Cl), 61.48 (CH2), 5.24 (CH3).
1-(5-(5-chloro-4-methyl-1-phenyl-1H-pyrazol-3-yl)-4-(8,8a-dihydro-[1,2,4]triazolo[5,1-c][1,2,4]triazin-7-yl)-3-oxocyclohex-1-en-1-yl)pyrimidine-2,4,6(1H,3H,5H)-trione (12)
Dissolve (0.012 mol) of 11 in xylene (20 ml), add formic acid (0.012 mol) then reflux the mixture for six hours. Once the solution is cooled down, filtration was performed of the separated solid, washed clean using water, dried down to remove moisture, and recrystallized using EtOH. Yield of 12 is 64% and melting point (mp) 286-287°C.
Analysis of 12 C24H20ClN9O4 (533) (%) Calculated C, 53.99; H, 3.76; Cl, 6.66; N, 23.61; Found: C, 54.00; H, 3.74; Cl, 6.65; N, 23.65.
FTIR spectra of 12 showed bands at 1620 cm-1, 1718,1735cm-1, 3340 cm1 for √ C=N, √ C=O, √ NH, absence of √ NH2. The ion peak of the solution was determined by mass spectrum and it was m/z 533(M+, 35.5Cl) (18.25%), 535 (M+, 37.5Cl) (11.23%). The 1H-NMR showed signal bands δ ppm at 10.06(s,1H, NH), 7.91(s,1H,NH triazole), 6.96-7.66(m,8H,ArH), 4.14(s,1H,CH=C), 3.04(d,1H,COCHCH), 3.31(q,1H, CHCHCH2), 3.25(d,2H, CH2CH), 3.08(s,2H,COCH2CO), 2.13(s,3H, CH3). 13C-NMR: 186.25,159.25, 155.35,151.02 (C=O), 162.25 (CN), 129.63, 127.05, 124.87, 122.09 C-aromatic, 138.99, 131.25(C=C alkene) 126.33(C-Cl), 5.09 (CH3).
1,1′-(4,4′-(5,5′-(2,2′-methylenebis(hydrazine-2,1-diyl))bis(4H-1,2,4-triazole-5,3-diyl))bis(5-(5-chloro-4-methyl-1-phenyl-1H-pyrazol-3-yl)-3-oxocyclohex-1-ene-4,1-diyl))dipyrimidine-2,4,6(1H,3H,5H)-trione (13)
Reflux for three hours (0.012 mol) formaldehyde and (0.012 mol) of compound 11, 3 ml of HCl. Once the solution cools down, filtration was performed of the separated solid, washed clean using water, dried down to remove moisture, and recrystallized from ethanol. Yield of 13 is 72% and melting point (mp) 310-311°C.
Analysis of 13 C45H40Cl2N18O8 (1030) (%) Calculated C, 52.83; H, 4.01; Cl, 7.22; N, 24.45; Found: C, 52.80; H, 4.13; Cl, 7.02; N, 24.44.
FTIR spectra of 13 showed bands at 1606 cm-1, 1690, 1725cm-1, 3342 cm-1 for √ C=N, √ C=O, √ NH respectively,absence of √ NH2. The ion peak of the solution was determined using a mass spectrum and it was m/z 1030(M+,35.5Cl) (2.06%), 1032(M+, 37.5Cl) (1.02%).
1-(4-(7H-[1,2,4]triazolo[1,5-d]tetrazol-6-yl)-5-(5-chloro-4-methyl-1-phenyl-1H-pyrazol-3-yl)-3-oxocyclohex-1-en-1-yl)pyrimidine-2,4,6(1H,3H,5H)-trione (14)
Stir (0.012 mol) of 11, sodium nitrite (0.012 mol), acetic acid (3.5 ml) in ethanol (25 ml) at 27°C for 4 h. Once the solution cooled down, filtration was performed of the separated solid, washed clean using water, dried down to remove moisture, and recrystallized from ethanol. Yield of 14 is 75% and melting point (mp) 275-276 °C.
Analysis of 14 C22H17ClN10O4 (520) (%) Calculated C, 50.73; H, 3.27; Cl, 7.12; N, 26.89; Found: C, 50.75; H, 3.29; Cl, 7.20; N, 26.88.
FTIR spectra of 14 showed bands at 1602 cm-1, 1725,1735 cm-1, 3320 cm1 for √ C=N, √ C=O, √ NH respectively, absence of √ NH2. The ion peak of the solution was noted using a mass spectrum and it was at m/z 520(M+, 35.5Cl) (4.36%), 522(M+, 37.5Cl), (2.25%). The 1H-NMR showed signal bands δppm at 10.24(s,2H,2x NH), 7.02-8.46(m,5H,ArH), 4.77 (s,1H,CH=C), 3.52(d,1H,COCHCH), 3.41(q,1H,CHCHCH2), 3.16(d,2H, CHCHCH2), 2.10(s,3H,CH3). 13C-NMR: 175.25, 156.75, 154.23, 151.98 (C=O), 141.53(CN), 128.65, 126.53, 124.86, 122.32 C-aromatic, 138.99, 131.25(C=C alkene), 124.56(C-Cl), 5.25 (CH3).
Biological Assessment
Anti-Diabetic Evaluation
Five new compounds, 1, 4b, 8, 12, and 14, were evaluated for their effect as inhibitors of NIDDM compared to the standard acarbose. α-Amylase and acarbose were used by the Anti-Diabetic Activity Division in RCMB (Regional Center for Mycology and Biotechnology). For the purpose of detecting the inhibitory activity, the derivative solution was tested in different concentrations (3.9, 7.8, 15.6, 31.25, 62.5, 125, 250, and 500 μg/mL). IC50 value is the value representation of the concentration of the new derivative which is needed to keep 50% inhibiting of α-amylase activity. The revelation from the outcomes is that the IC50 of the control was 12.87 µg and that compounds 4b, 12, and 14 showed IC50 values of 13.54, 17.08, and 21.359 μg/ml, respectively. The high potency of these compounds can be explained by the structure-activity relationship, where the reactivity of compound 4b is attributed to the presence of a thione group. Thus, the derivatives bearing a strong electron-withdrawing group showed potent activity. Likewise, the reactivity of compounds 12 and 14 is attributed to the triazine moiety fused with tetrazoles.
Table 1: Anti-diabetic activity of tested compounds.
|
Alpha-glucosidase inhibitory |
||||||
|
Sample conc. (µg) |
12 |
4b |
14 |
1 |
8 |
Reference (control) Acarbose |
|
500 |
76.32±1.20 |
88.32±0.63 |
80.41±1.5 |
79.35±0.63 |
79.31±2.5 |
94.35±1.20 |
|
250 |
70.35±0.72 |
81.35±1.50 |
74.35±1.2 |
71.34±1.5 |
69.32±2.1 |
86.47±0.58 |
|
125 |
68.24±0.58 |
72.36±1.20 |
69.31±201 |
70.32±3.1 |
61.34±0.72 |
81.32±0.63 |
|
62.50 |
61.38±0.63 |
64.35±0.58 |
58.37±.58 |
60.34±0.58 |
59.37±0.58 |
74.15±0.72 |
|
31.25 |
56.84±1.20 |
58.32±0.63 |
50.42±0.63 |
42.58±0.72 |
54.34±0.58 |
63.25±1.50 |
|
15.63 |
49.32±0.58 |
50.87±0.72 |
41.38±0.72 |
36.74±1.5 |
43.28±1.5 |
52.00±0.58 |
|
7.81 |
39.28±0.72 |
42.15±1.20 |
39.25±1.5 |
24.74±1.2 |
32.58±1.2 |
46.31±0.63 |
|
3.90 |
31.25±1.20 |
29.54±0.72 |
21.35±0.72 |
0 |
30.14±3.1 |
36.21±1.20 |
|
0 |
0 |
0 |
0 |
0 |
0 |
0 |
|
IC50 |
17.04µg |
13.80µg |
21.35 µg |
44.37 µg |
25.09 µg |
12.87µg |
Anti-Cancer Evaluation
The anti-tumor cytotoxicity of compounds 1, 2a, 4b, 5, 6, 9, 10, 11, and 14 was assessed and compared to that of doxorubicin as a reference; a total of 3 types of cancer cell lines were tested, namely: HCT116, HEPG2, and MCF7. IC50 values and cytotoxicity values of each solution were tested as shown in (table 2-4). With an increase in the concentration of the sample compound, a gradual decrement was detected in the survival fraction. As shown in the results table, in the samples 10, 6, 4a, and 11, the compounds scored the lowest values for IC50, which is a signifier that these compounds serve as the highest potent cytotoxic drugs. The high potency of these compounds can be explained by the structure-activity relationship, where the reactivity of compound 4b is attributed to the presence of a thione group that takes advantage of the electron-withdrawing group substituted on the pyrimidine ring, that of compound 6 is credited to involvement of an element belonging to a carbonitrile group, and that of compounds 10 and 11 is attributed to the presence of a cyclohexene moiety.
Table 2: Cytotoxicity inhibition of compounds 1,5,11 and Doxorubicin against hepatocellular carcinoma cell line.
|
Conc. µg/mL |
Viability (%) / compound |
|||
|
1 |
5 |
11 |
Doxorubicin |
|
|
50 |
16.19 |
26.46 |
8.15 |
10.86 |
|
25 |
68.47 |
77.33 |
36.93 |
23.18 |
|
12.5 |
79.68 |
84.86 |
65.93 |
39.18 |
|
6.25 |
89.14 |
93.17 |
82.14 |
50.25 |
|
3.125 |
96.42 |
98.65 |
93.42 |
74.56 |
|
1.56 |
100 |
100 |
98.57 |
90.82 |
|
0 |
100 |
100 |
100 |
100 |
|
IC50 |
33.8 |
33 |
19.3 |
9 |
Table 3: Cytotoxicity inhibition of compounds 2a,4b,9 and Doxorubicin against breast carcinoma cell line.
|
Conc. µg/mL |
Viability (%) / compound | |||
|
2a |
4b |
9 |
Doxorubicin |
|
|
50 |
60.56 |
4.35 |
23.67 |
6.14 |
|
25 |
78.42 |
9.62 |
58.04 |
10.98 |
|
12.5 |
88.35 |
13.37 |
80.43 |
16.86 |
|
6.25 |
94.77 |
26.41 |
92.54 |
25.94 |
|
3.125 |
98.48 |
37.23 |
98.16 |
34.63 |
|
1.56 |
100 |
74.23 |
100 |
67.91 |
|
0 |
100 |
100 |
100 |
100 |
|
IC50 |
≥50 |
2.6 |
30.8 |
2.4 |
Table 4: Cytotoxicity inhibition of compounds 6,10,14 and Doxorubicin against colon carcinoma cell line.
|
Conc. µg/mL |
Viability (%) / compound |
|||
|
6 |
10 |
14 |
Doxorubicin |
|
|
50 |
7.18 |
5.8 |
8.36 |
5.43 |
|
25 |
12.94 |
10.26 |
14.27 |
9.72 |
|
12.5 |
28.89 |
17.93 |
26.14 |
17.25 |
|
6.25 |
56.79 |
41.12 |
69.68 |
28.02 |
|
3.125 |
72.58 |
63.77 |
84.71 |
57.18 |
|
1.56 |
90.64 |
80.98 |
92.23 |
76.72 |
|
0 |
100 |
100 |
100 |
100 |
|
IC50 |
7.7 |
5 |
9.1 |
3.9 |
Result and Discussion
Synthesis and Characterization
Condensation of barbituric acid via Knoevenagel condensation through the reactive methylene group within the 5-chloro-4-methyl-1-phenyl-1H-pyrazole-3-carbaldehyde moiety, which had been prepared by the conventional methods,17 afforded the corresponding α, β-unsaturated pyrazol-3-ylmethylenepyrimidine-2,4,6-trione, which leads to a new heterocyclic derivatives, as shown in Scheme 1. This new chalcone undergoes nucleophilic cycloaddition after treatment with hydrazine hydrate or phenyl hydrazine in boiling ethyl alcohol, causing production of 2a and b, as indicated by the absence of a signal at δ 6.52 ppm attributed to C=CH in the 1H-NMR spectrum. Additionally, the 13C-NMR spectrum (DMSO-d6) for 2a showed bands at 158.96 and 155.05 (C=O), 140.05 and 138.55 (C=N); 134.07 and 129.08 (C=C); 133.25 (C-N); 128.68, 125.54, and 123.43 (C-aromatic); 124.98 (C-Cl); and 9.06 (CH3).
Chalcone undergoes cycloaddition with hydroxyl amine in boiling pyridine to afford an isoxazolo moiety; this ring formation is confirmed by spectral data, with the following signals observed in the 13C-NMR spectrum (DMSO-d6): 154.96 and 152.45 (C=O); 149.98 (C=N); 129.46, 126.54, and 123.66 (C-aromatic); 128.23 and 127.75 (C=CH); 125.88 (C-Cl); and 6.16 (CH3).
On the other hand, the 1,3-dipolar cyclocondensation of 1 with different carbamides, namely, urea, thiourea and guanidine HCl, in glacial acetic acid afforded derivatives 4a, b, and c. The formation of the six-membered pyrimidine ring was confirmed by the spectral data; for example, compound 4b resulted in the IR spectrum with a clear absorption band at 1410 cm-1 for νC=S. Additionally, the 13C-NMR spectrum showed signals at δ (ppm) 139.75 and 138.05 (C=N), 135.07 and 132.18 (C=C), and 129.85 (C-N), which proved the formation of the pyrimidine ring.
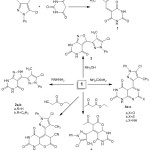 |
Scheme 1: Synthesis of compounds 1, 2(a-b), 3, 4(a-c), 5 and 6. |
On the other hand, condensation of chalcone with ethyl acetoacetate or ethyl cyanoacetate via the reactive methylene group followed by elimination of ethanol or water, respectively, and then cyclization to form compounds 5 and 6, respectively. The identity of compound 5 was characterized by spectral data, FTIR results showed bands at 1210 cm-1 for νC-O, indicating the formation of a pyrano ring. The 1H-NMR also showed singlet bands at δ 3.75 and 2.37 ppm corresponding to CHCO, COCH3,in the same order.
The 13C-NMR spectrum showed signals at 159.26, 153.85, and 151.75 (C=O); 139. 98 (C=N); 128.26, 125.84, and 122.76 (C-aromatic); 128.23 and 127.75 (C=CH); 125.88 (C-Cl); and 6.16 and 28.01 (CH3). Moreover, FTIR spectra of 6 showed band at 2234 cm-1 for νC≡N. The 1H-NMR showed singlet bands at δ 5.03 and 4.24 ppm corresponding to CH-O and CHCN, respectively, which proved the formation of a six-membered ring.
The acetylation of barbituric acid with acetyl chloride in a water bath afforded 7. This acetylation occurred on only the NH group, while the other group gave rise to a singlet signal in the 1H-NMR spectrum at δ 10.22 ppm. This acetylated derivative is one of the initial materials use to synthesize the new compounds, as shown in Scheme 2.
Condensation of 7 with pyrazole-3-carbaldehyde in ethanol and 10% sodium hydroxide yielded the pyrazol-3-yl(acryloyl)pyrimidine-2,4,6-trione derivative 8; although condensation was thought to occur at the methylene group of the pyrimidinone ring, an occurrence of a singlet band at δ 3.03 ppm in the 1H-NMR spectrum and the absence of a singlet band for NHCOCH3 indicated that the condensation occurred at the methyl group.18
The identity of compound 8 was chemically confirmed by treating 8 with the dicarbonyl compound ethyl acetoacetate in ethanol and sodium ethoxide to yield cyclohexanone-3-enecarboxylate 9, which is consistent with the data from the IR spectrum showing absorption bands at 1190 cm-1 for νC-O. On the 1H-NMR spectrum, there was a quartet signal at δ 3.65 alongside a triplet band at 1.45 for COCH2CH3 of the carboxylate group. The formation of the cyclohexenone group was also indicated by 1H-NMR, which showed a quartet signal at δ 3.44 attributed to CHCHCH2 and doublet signals at 3.28 and 3.22 ppm that correspond to CHCHCH2.
The nucleophilic addition of thiosemicarbazide to 9 in presence of sodium ethoxide produced compound10, as confirmed by the spectral data showing an absorption band at 1410 cm-1 refer to νC=S. The reaction of 10 with hydrazine hydrate compound in boiling ethanol gave 11. On the IR spectrum, there was no reading of an absorption band forming, which can be explained through νC=S and stretching bands at 3330 and 3340 cm-1 attributable to νNH2. Additionally, the formation of the hydrazinyl group was indicated via 1H-NMR, which showed a triplet signal at 4.89 ppm corresponding to NHNH2 and a doublet at 2.55 ppm corresponding to NHNH2.
This new hydrazinyl derivative was chemically confirmed using a formic acid treatment in boiling ethanol to form sample compound 12. The reaction was carried out through formation of the hydrazinyl group followed by cyclization. The spectral data revealed the absence of NH2 and the formation of the triazine ring.
Treatment of 11 with formaldehyde in boiling HCl afforded dimer 12. This dimer was thought to be formed via condensation between 2 mol of the hydrazinyl derivative and formaldehyde. The expected structure agrees with spectral data, the ion peak of the solution was noted using a mass spectrum and it was m/z 1030 (M+, 35.5Cl) (2.06%).
The hydrazinyl derivative converted into 14 via reaction with nitrous acid through diazotization of the hydrazinyl group. The formation of the tetrazole derivative is confirmed by spectral data. Data collected using spectral analysis and elemental analysis was used to prove the structured of all the newly synthesized compounds.
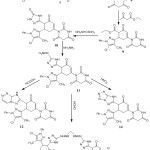 |
Scheme 2: Synthesis of compounds 7-14. |
α-Amylase Assay
In order to measure the inhibition efficiency of the compounds in α-amylase, the protocol defined by McCue19 was used with some minor changes to fit the experiment adequately. For the purpose of determining the α-amylase inhibitory activity, each compound was tested using different concentration samples in methanol. For the testing, each fraction was taken in 250 micro-liter in volume or varying concentrations (from 30 to 240 micro-liter/ml) of acarbose. This mixture was then subjected to incubation for twenty minutes at 37 degree-Celsius in a porcine pancreatic amylase (2 U/ml) in a buffer of phosphate (100 mM and pH value of 6.8). Afterwards, 250 micro-liter of 1% starch dissolved in the buffer of 100 mM phosphate (pH value of 6.8) is also added to the aforementioned mixture. The resulting solution is subjected to incubation for one hour at temperature of 37 degree-Celsius. This mixture is then boiled for a duration of ten minutes after adding a color reagent in the form of Dinitrosalicylate. For the final mixture compound, the value of absorbance is noted at 540 nm. In this procedure of α-amylase, the standard benchmark was Acarbose. To describe the inhibitory potential of the compound in α-amylase, a percentage value is represented that is calculated using the following formula:
![]()
Where At = the absorbance of the compound tested.
Ac = the absorbance of control.
Cytotoxic Activity
The new derivatives were tested against human cancer cell lines at the Anti-Tumor Activity Unit in the RCMB. Three different cancer cell lines, MCF7, HEPG2, and HCT 116 were procured in a frozen state submerged inside liquid nitrogen from the American Type Culture Collection. Serial sub-culturing method was used to maintain the tumor cell lines. The potential cytotoxicity of 1, 2a, 4b, 5, 6, 9, 10, 11, and 14, as well as doxorubicin, was validated using the method described by Skehan et al.20 The relationship of the drug concentrations in µg/mL was plotted against the cell viability to obtain the survival fraction IC50 values of the new derivatives.
Conclusion
Conversion of barbituric acid into new heterocyclic compounds results in the formation of a new series of bioactive molecules containing the pyrimidine nucleus incorporated with pyrazolyl, pyrimidone, isoxazole, pyranone, and triazole moieties. Data collected using spectral analysis and elemental analysis was used to prove the structured of all the newly synthesized compounds. The anti-diabetic reactivity of some of the synthesized compounds was verified in vitro. Some compounds showed high inhibition activity, and compound 4b was the most active (IC50 13.54 µg/ml), with an activity very close to that of the standard acarbose. The structure-activity relationship of these compounds was studied, and the reactivity of compound 4b is attributed to the presence of a thione group.
Compound 4b (IC50 2.6 µg/ml) has the potential use in the form of a cytotoxic drug for treatment of breast carcinoma cells. 11 (IC50 19.3 µg/ml) can be used for liver carcinoma cells, and 6 and 10 (IC50 7.7, 5 µg/ml) can be used as colon carcinoma cell cytotoxic drugs.
Conflicts of Interest
Author hasn’t any conflicts of interest.
Acknowledgements
Facilities provided by Imam Abdulrahman Bin Faisal University and the Regional Center for Mycology and Biotechnology are gratefully acknowledged.
References
- Felczak, K.; Bretner, M.; Kulikowski, T.; Shugar, D. Nucleosides Nucleotides 1993, 12(2), 245-261.
- Sharma, V.; Chitranshi, N.; Agarwal, A.K. Int. J. Med. Chem. 2014, 202784.
- Imran, M.; Khan, S.A. Trop. J. Pharm. Res. 2015, 14(7), 1265-1272.
- UI-Haq, Z.; Ashraf, S.; Al-Majid, A.M; Barakat, A. Int. J. Mol. Sci. 2016, 17(5), 657.
- Ma, L.; Li, S.; Zheng, H.; Chen, J.; Lin, L.; Ye, X.; Chen, Z.; Xu, Q.; Chen, T.; Yang, J.; Qiu, N.; Wang, G.; Peng, A.; Ding, Y.; Wei, Y.; Chen, L. Eur. J. Med. Chem. 2011, 46(6), 2003–2010.
- Faidallah, H.M.; Khan, K.A. J. Fluor. Chem. 2012, 142, 96-104.
- Dixit, V.A.; Rathi, P.C.; Bhagat, S.; Gohlke, H.; Petersen, R.K.; Kristiansen, K.; Chakraborti, A. K.; Bharatam, P.V. Eur. J. Med. Chem. 2016, 108, 423-435.
- Moussier, N.; Bruche, L.; Viani, F.; Zanda, M. Curr. Org. Chem. 2003, 7(11), 1071–1080.
- Khodeza Khatun, M.; Al-Reza, S.M.; Sattar, M.A. Int. J. Adv. Res. Chem. Sci. 2016, 3(6), 21–26.
- Tanaka, K.; Chen, X.; Yoneda, F. Tetrahedron 1988, 44(11), 3241–3249.
- Sokmen, B.B.; Ugras, S.; Sarikaya, H.Y.; Ugras, H.I.; Yanardag, R. Appl. Biochem. Biotechnol. 2013, 171(8), 2030-2039.
- Yan, Q.; Cao, R.; Yi, W.; Chen, Z.; Wen, H.; Ma, L. Eur. J. Med. Chem. 2009, 44(10), 4235–4243.
- Nabil, S.; Abd El-Rahman, S.N.; Al-Jameel, S.S.; Elsharif, A.M. Biological and pharmaceutical Bulletin 2018, 41(7), 1071-1077.
- Mohamed, M.I.; Zaky, H.T.; Kandile, N.G. J. Chinese Chemical Society 2004, 51, 963-968.
- Zhang, P.; Wei, C.; Wang, E.; Wang, W.; Liu, Q.; Chen, H.; Wang, K. J. Carbohydr. Res. 2012, 351, 7–16.
- Das, M.; Manna, K. Current Bioactive Compounds 2015, 11(4), 239–248.
- Abd, E.; Al, S.N.; Soliman, F.M.A. Der Pharma Chemica 2015, 7(4), 71-84.
- Aly, H.M.; Saleh, N.M.; Elhady, H.A. Eur. J. Med. Chem. 2011, 46(9), 4566–4572.
- McCue, P.P.; Shetty, K. I. Asia Pac. J. Clin. Nutr. 2004, 13(1), 101-6.
- Skehan, P.; Storeng, R.; Scudiero, D.; Monks, A.; McMahon, J.; Vistica, D.; Warren, J.T.; Bokesch, H.; Kenney, S.; Boyd, M.R. J. Natl. Cancer Inst. 1990, 82(13), 1107–1112.

This work is licensed under a Creative Commons Attribution 4.0 International License.
![]()
A New Edition of Web of Science
Journal Impact Factor
2022: 0.5
Five Year: 0.8
Journal is Indexed in

Cabells Whitelist
![]()


