Theoretical Study of Reaction Between Nitrilimine and 1,4 Oxazine 2 Carboxylate by MP2 and DFT Methods
H. El-Hadki1 , F. Hlimi2, M. Salah1, K. Marakchi1, N. Komiha1 and O. K. Kabbaj1
, F. Hlimi2, M. Salah1, K. Marakchi1, N. Komiha1 and O. K. Kabbaj1
1CERNE2D; Laboratory of Spectroscopy Molecular Modeling Materials Nanomaterials water and Environment (LS3MN2E) - Faculty of Sciences-University Mohammed V-Rabat-Morocco.
2Laboratory of plant chemistry and organic and bioorganic synthesis (LCPSOB) - Faculty of Sciences-University Mohammed V-Rabat-Morocco.
Corresponding Author E-mail: hamza.hadk@gmail.com
DOI : http://dx.doi.org/10.13005/ojc/340640
Article Received on : 20-07-2018
Article Accepted on : 11-11-2018
Article Published : 15 Nov 2018
The regioselectivity of the reaction between phenyldiazen-1-ium-1-ylidene p-tolyl methanide and ethyl 4-benzoyl-4H-benzo [1,4]oxazine-2-carboxylate were studied by means of the DFT/B3LYP method coupled by the 6-31g(d) basis and MP2 in connection with 6-31G(d) and 6-31G+(d,p) basis set. The mechanism of this regioselectif reaction has performed by transition state optimization, evaluation of the potential energy surface, calculation of IRC and reactivity indices. Location and verification of minima and transition structures have been realized by using the Berny's algorithm. The process of formation of the two regioisomers is achieved through concerted and asynchronous mechanism. The results are in good agreement with the experimental data.
KEYWORDS:Carboxylate; Global Reactivity Descriptors; Pyrazoline; Nitrilimine; Quantum Chemical Calculations
Download this article as:| Copy the following to cite this article: El-Hadki H, Hlimi F, Salah M, Marakchi K, Komiha N, Kabbaj O. K. Theoretical Study of Reaction Between Nitrilimine and 1,4 Oxazine 2 Carboxylate by MP2 and DFT Methods. Orient J Chem 2018;34(6). |
| Copy the following to cite this URL: El-Hadki H, Hlimi F, Salah M, Marakchi K, Komiha N, Kabbaj O. K. Theoretical Study of Reaction Between Nitrilimine and 1,4 Oxazine 2 Carboxylate by MP2 and DFT Methods. Orient J Chem 2018;34(6). Available from: http://www.orientjchem.org/?p=52640 |
Introduction
1,3 dipolar cycloadditions of nitrilimine with 1,4 oxazine 2 carboxylate is a very powerful tools for the synthesis of a large range of pyrazoline derivatives that occupy an important place in medicinal and pesticide chemistry.They present a wide range of bioactivity such as anti-inflammatory,1 analgesic,2 antagonist,3 anti-depressant and anti-convulsant,4 antitumour,5 antioxidant,6 and antimicrobial.7 The large field of biological activities of pyrazolines has made them widespread synthetic targets.Extensive research efforts are continually directed at the discovery of new heterocycles with appropriate pharmacological effects. It is admitted that the most popular method for synthesis of pyrazolines is 1,3-dipolar cycloaddition of diazo compounds to electron deficient alkenes.8-10 Several previous studies have been carried out on the reactions of Cycloaddition 1, 3-dipole are reported in the literature.11-14 Hlimi and collaborators15 studied experimentally the reaction between N-benzoyl benzoxazine-1.4 carboxylate-2 ethyl (A) and C-P-tolyl N-Phenylnitrilimine (B) (Figure 1) and concluded that this reaction is regiospecific with the formation of ortho regioisomer.
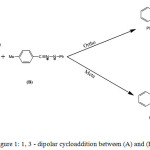 |
Figure 1: 1, 3 – dipolar cycloaddition between (A) and (B). |
To better understand this reaction, we have studied and discussed in detail his mechanism by using DFT-based conceptual indices of reactivity and stationary point analysis on potential energy surfaces on both tracts. This analysis clarifies the regioselectivity of this reaction and explains the experimental results.
Materials and Methods
Theoretical approaches based on the MP2 and DFT methods stay strong tools to rationalize the chemical reactivity and selectivity of reaction, object of study. The calculations were performed with the software GAUSSIAN0916 and visualized by the graphical interface Gauss View Package.17 Structures optimizations have been computed using B3LYP/6-31G(d) without symmetry constraint then a single point calculation was using MP2/6–31G(d) and MP2/6-31G+(d,p). Harmonic vibration frequencies were determined to confirm local minimum on the potential energy surface. In the reaction mechanism, we determined the presence of two transition states. For each one, we traced the IRC’s paths to authenticate the connection between transition states (TS) and associated minima. The structures of compounds were analyzed by NBO method.18-20 In order to reveal the molecular reactivity and electronic properties of different stationary point, we have focused at the energies of frontier molecular orbitals (FMO’s) to determine global indices: electronic chemical potential (µ), hardness (η), global electrophilicity (ω) and global nucleophilicity (N) calculated using the following expressions:
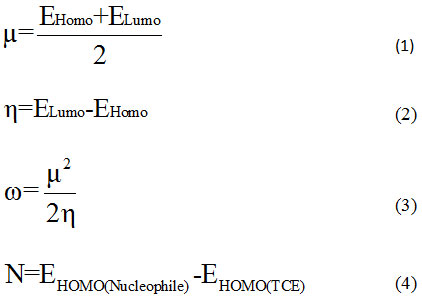
where EHomo and ELumo are the energies of frontier molecular orbitals. The global nucleophilicity N is assign to tetracyanoethylene (TCE)21-22 who has lowest HOMO energy and very large electrophilicity.
The use of the local index of Fukui (ωk) allows the prediction of the formation of the most favorable binding. They are respectively indicated by the expressions:

fk+ and fk– are the condensed forms of the functions of Fukui to electrophilic and nucleophilic attacks,
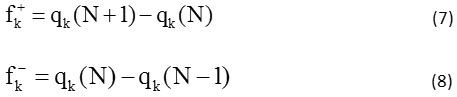
where qk (N), qk (N+1), and qk (N-1) represent respectively the charge on the kth atom in neutral, cationic and anionic chemical species.
Toro-Labbé et al.,23-24 suggests a dual descriptor Δfk for specifying the site reaction of the compound is given as follow:
![]()
The multiphilic descriptor Δωk, index of selectivity towards nucleophilic attack, is given according to the following expression:
![]()
Results and Discussion
Global Reactivity Indices
The results for the global reactivity indices are grouped in Table 1.
Table 1: Energies of HOMO and LUMO, global reactivity descriptor in eV.
| Methods | Compound | E(Homo) | E(Lumo) | μ | η | ω | N |
| DFT/B3LYP/6-31G(d) | A | -5,44 | -1,47 | -3,46 | 3,97 | 1,51 | 3,68 |
| MP2/6-31G(d) | -7,92 | 2,30 | -2,81 | 10,22 | 0,39 | 3,79 | |
| MP2/6-31+G(d,p) | -8,11 | 1,55 | -3,28 | 9,66 | 0,56 | 3,75 | |
| DFT/B3LYP/6-31G(d) | B | -4,89 | -1,33 | -3,11 | 3,56 | 1,36 | 4,23 |
| MP2/6-31G(d) | -6,65 | 1,96 | -2,34 | 8,61 | 0,32 | 5,06 | |
| MP2/6-31+G(d,p) | -6,84 | 1,51 | -2,67 | 8,35 | 0,43 | 5,02 |
The analysis of the results obtained shows that the compounds A and B play respectively the role of dipolarophile and dipole; indeed μ (A) < μ (B) and N(A) < N(B). It can therefore be concluded that the studied reaction has a normal electronic demand (NED).
Prediction of Regiochemistry using the Dual and the Multiphilic Descriptor
To predict the site of a nucleophilic and electrophilic attack, we have used the dual descriptor Δfk and multiphilic descriptor Δωk. The calculations led to the results reported in table 2.
Table 2: Multiphilic Descriptor Δf, Δω of atoms C1, C2 of dipolarophile and N1, N3 of dipole with NPA analysis.
| Methods | Compound | fk+ | fk– | Δfk | ω k+ | ωk– | Δωk | |
| DFT/B3LYP/6-31G(d) | Dipole | C3 | -0,021 | 0,154 | -0,175 | -0,029 | -0,237 | 0,209 |
| N1 | -0,075 | 0,111 | -0,186 | -0,102 | -0,253 | 0,151 | ||
| MP2/6-31G(d) | C3 | 0,238 | 0,246 | -0,009 | 0,076 | -0,003 | 0,079 | |
| N1 | 0,366 | 0,285 | 0,081 | 0,117 | 0,026 | 0,091 | ||
| MP2/6-31+G(d,p) | C3 | 0,233 | 0,253 | -0,02 | 0,099 | -0,008 | 0,107 | |
| N1 | 0,389 | 0,322 | 0,068 | 0,166 | 0,029 | 0,137 | ||
| DFT/B3LYP/6-31G(d) | Dipolarophile | C1 | -0,067 | 0,013 | -0,08 | -0,101 | -0,109 | 0,008 |
| C2 | -0,069 | -0,022 | -0,046 | -0,103 | -0,063 | -0,041 | ||
| MP2/6-31G(d) | C1 | 0,063 | -0,018 | 0,081 | 0,024 | 0,031 | -0,007 | |
| C2 | 0,045 | 0,1 | -0,054 | 0,018 | -0,021 | 0,039 | ||
| MP2/6-31+G(d,p) | C1 | 0,054 | -0,025 | 0,079 | 0,03 | 0,044 | -0,014 | |
| C2 | 0,039 | 0,099 | -0,06 | 0,022 | -0,034 | 0,055 | ||
According to the results of DFT method (Table 2), the positive value of Δωk (0,21) of the atom C3 of the dipole indicates that it’s the most favorable centre for nucleophilic attack. Regarding to the dipolarophile, atom C2 will undergo an electrophilic attack (negative value of Δωk (-0,04). Therefore, the first formed bond will be between atoms C2 of dipolarophile and C3 of dipole. The second formed bond will take place between the atoms N1 of dipole and C1 of dipolarophile. The obtained results are in good agreement with experimental study.15 On the other hand, the results obtained with the MP2 method lead first to the formation of the N1C1 bond followed by the C2C3 bond.
The two methodological approaches allowed us to understand and interpret the results obtained experimentally i.e. the formation of the regioisomer in position ortho as can be seen below:
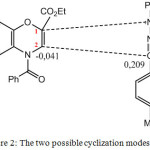 |
Figure 2: The two possible cyclization modes. |
Mechanistic study
Determination of TS’s
In the following, we chose to determine the reaction profile using DFT method since MP2 leads to the same result.
Theoretical predictions of the regioselectivity will be based on the transition state. The reaction between the two compounds A and B leads to two products P1 and P2 respectively characterizing the two regioisomers ortho and meta through single transition structures on each reactionels paths (TS1 or TS2). The optimized geometries of ortho and meta transition states are reported in the figure 3.
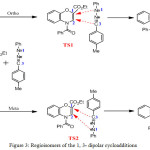 |
Figure 3: Regioisomers of the 1, 3- dipolar cycloadditions. |
The total energy of all studied compounds are summarized in table 3 and figure 4
Table 3: Total energies and relative energies of reactants, products and TS’s.
| E (u.a) | DE (Kj/mol) | ||
| Reactives | Dipole | -65,014,198,108 | |
| Dipolarophile | -105,059,332,848 | ||
| Ortho-pathway | TS 1 | -170,071,422,962 | 55,3 |
| P1 | -170,079,311,216 | -151,8 | |
| Meta-pathway | TS 2 | -170,072,012,043 | 39,9 |
| P2 | -170,079,261,930 | -150,5 | |
 |
Figure 4: Relative energies of transition states and products. |
The values of the activation barrier between the dipole (nitrilimine) and the dipolarophile are 55,3 Kj/mol for TS1 and 39,9 Kj/mol for TS2, consequently the P2 is the regioisomer kinetically favored to be formed. Thermodynamically we can’t determine the favored regioisomer because the variation of energy between P1 and P2 is low in the order of 1,3 KJ/mol.
Optimized Geometries of TS’s
Geometries of TS’s corresponding to our reaction are depicted in table 4 and figure 5.
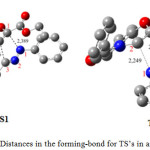 |
Figure 5: Distances in the forming-bond for TS’s in angstroms. |
The corresponding selected geometric parameters reveal that the transition structures are asynchronous. The degree of asynchronicity Dd = d (N-C) – d(C-C) determined by difference between the two forming bonds is regrouped in table 4.
Table 4: Degree of asynchronicity for the two pathways.
| Ortho-pathway | Meta-pathway | ||||
| d(N1-C1) | d(C3-C2) | Δd | d(N1-C2) | d(C3-C1) | Δd |
| 2.389 | 2.186 | 0.203 | 2.249 | 2.335 | 0.086 |
The difference for TS1 (0.203 Å) and TS2 (0.086 Å) seems to indicate that the formation of the TS1 is asynschrone, while for the TS2 the mechanism of formation take place with a synchronous process.
Transition Vectors and Frequencies Analysis
In order to understand the process related to each transition structure, an analysis of the transition vectors (TVs) is performed. Imaginary frequencies, TVs components and geometric parameters are reported in table 5.
Table 5: Imaginary frequency, TVs eigenvalues for the pathways.
| Ortho-pathway | Meta-pathway | ||||
| d(N1-C1) | d(C3-C2) | Δd | d(N1-C2) | d(C3-C1) | Δd |
| 2.389 | 2.186 | 0.203 | 2.249 | 2.335 | 0.086 |
The dominant components for the ortho-pathway are C3-C2 and N1-C1 respectively associated to 0.52 and 0.33 a.u corresponding to s-bonds newly formed. For the other pathway, the associated values are C3-C1 (0.64) and N1-C2 (0.30). These data confirm that the studied reaction is asynchronous.
Charge Transfer in TSs
NBO (Natural bond orbital) theory has allowed us to determine the natural loads between the dipole and the dipolarophile and calculate the global electron density transfer (GEDT).25-26 The results are listed in table 6.
Table 6: Global electron density transfer in eV using DFT/B3LYP/6-31G(d).
| GEDT | TS1 | TS2 |
| 0.082 | 0.024 |
The positive value indicates a flow of Electron density of the nucleophile to the electrophilic, this confirms the results obtained previously for the chemical potential.
Conclusion
In this work, we studied the reaction of cycloaddition between C-P-tolyl N-Phenylnitrilimine (dipole) and N-benzoyl benzoxazine-1.4 carboxylate-2 ethyl (dipolarophile) exploring the potential energy surface and using the descriptors of the multiphilic at the level of the DFT/B3LYP/6-31 G (d), MP2/6-31G(d) and MP2/6-31G+(d,p). The two methods used for the determination of the analysis of electronic chemical potentials, hardness at reactive sites of dipole and dipolarophile indicate a normal demand character.
We have reproduced the two reaction pathways leading to both ortho and meta regioisomers; the results led to the privileged formation of ortho regioisomer. The analysis of transition states reveal that ortho regioisomer takes place through concerted mechanism with asynchronous character while more or less synchronous character for the meta pathway. This theoretical study confirms the experimental data of the team of Hlimi which shows that majoritory product is phenyl-1 p-tolyl-3 benzoyl-4 dihydro-3a, 9 a pyrazolo [4,5-6] benzoxazine l,4 carboxylate-9a ethyl
Acknowledgements
The authors thank AMCT for Gaussian program.
Reference
- Bandgar, B.P.; Gawande, S.S.; Bodade, R.G.; Gawande, N.M.; Khobragade, C.N.Bioorganic & Medicinal Chemistry. 2009, 17, 8168‑8173.
CrossRef - A. Gürsoy, A.; Demirayak, Ş.; Çapan, G.; Erol, K.; Vural, K. European Journal of Medicinal Chemistry.2000, 35, 359‑364.
CrossRef - McKeown, S.C.; Hall, H.; Giblin, G. M. P.; Lorthioir, O.; Blunt, R.; Lewell, X. Q.; Wilson, R.J.; Brown, S. H.; Chowdhury, A.; Coleman, T.; Watson, S.P.; Chessell, I.P.; Pipe, A.; Clayton, N.; Goldsmith, P. Bioorganic & Medicinal Chemistry Letters. 2006, 16, 4767-4771
CrossRef - Özdemir, Z.; Kandilci, H. B.; Gümüşel, B.; Çalış, Ü.; Bilgin, A. A.European Journal of Medicinal Chemistry.2007,42, 373‑379.
CrossRef - Soni, H.M.; Patel, P.K.; Chhabria, M.T.; Patel, A.K.; Rana, D.N.; Brahmkshatriya, P. S.International Journal of Organic Chemistry.2016, 06, 157‑176.
CrossRef - Govindaraju, M.; Govindappa, V. K.; Pavithra, G.; Nayaka, M.; Ningappa, M.; Kumar, K. Journal of Pharmacy and Biological Sciences (IOSRJPBS). 2012, 2,2278‑3008.
- Kumar, G. V.; Govindaraju, M.; Renuka, N.; Khatoon, B. B. A.; Mylarappa, B. N.; Kumar, K. A. RASAYAN J. Chem. 2012, 5, 338-342.
- Padwa, A.; Woolhouse, A.D. Comprehensive Heterocyclic Chemistry, 1984, 7, 47-93
- Zhang, Z.; Wang, J.Tetrahedron. 2008, 64, 6577‑6605.
CrossRef - Domingo, L. R.; Ríos-Gutiérrez, M.; Emamian, S. RSC Advances. 2017, 7, 15586‑15595.
CrossRef - Molteni, G.; Ponti, A.Molecules. 2017, 22,202-214.
CrossRef - Bakavoli, M.; Moeinpour, F.; Davoodnia, A.; Morsali,A. Journal of Molecular Structure. 2010, 969, 139‑144.
CrossRef - Moeinpour, F. Chinese Journal of Chemical Physics. 2010, 23, 165-168
CrossRef - Moeinpour,F.; Khojastehnezhad, A.Acta chimica Slovenica. 2015, 62, 403‑410.
CrossRef - Hlimi, F.; Kerbal, A.; Laude, B.; Vebrel, J. C. R. Acad. Sci. Paris, 1988, t. 306, Série II, 1437-1440,
- Frisch, M. J.; Trucks, G. W.; Schlegel, H. B.; Scuseria, G. E.; Robb, M. A.; Cheeseman, J. R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G. A.; Nakatsuji, H.; Caricato, M.; Li, X.; Hratchian, H. P.; Izmaylov, A. F.; Bloino, J.; Zheng, G.; Sonnenberg, J. L.; Hada, M.; Ehara, M.; Toyota, K.; Fukuda, R.; Hasegawa, J.; Ishida, M.; Nakajima, T.; Honda, Y.; Kitao, O.; Nakai, H.; Vreven, T.; Montgomery, J. A.; Peralta, Jr. J. E.; Ogliaro, F.; Bearpark, M.; Heyd, J. J.; Brothers, E.; Kudin, K. N.; Staroverov, V. N.; Kobayashi, R.; Normand, J.; Raghavachari, K.; Rendell, A.; Burant, J. C.; Iyengar, S. S.; Tomasi, J.; Cossi, M.; Rega, N.; Millam, J. M.; Klene, M.; Knox, J. E.; Cross, J. B.; Bakken, V.; Adamo, C.; Jaramillo, J.; Gomperts, R.; Stratmann, R. E.; Yazyev, O.; Austin, A. J.; Cammi, R.; Pomelli, C.; Ochterski, J. W.; Martin, R. L.; Morokuma, K.; Zakrzewski, V. G.; Voth, G. A.; Salvador, P.; Dannenberg, J. J.; Dapprich, S.; Daniels, A. D.; Farkas, O.; Foresman, J. B.; Ortiz, J. V.; Cioslowski, J.; Fox, D. J. 2009, Gaussian 09, Revision A.02, Gaussian, Inc., Wallingford CT.
- Dennington II, R.; Keith, T.; Millam, J.; Eppinnett, K.; Hovell, W.L.; Gilliland, R.; Gauss View, Version 3.0.9, Semichem Inc., Shawnee Mission, KS, 2003.
- Keresztury, G.; Holly, S.; Besenyei, G.; Varga, J.; Wang, A.; Durig, J. R.Spectrochimica Acta Part A: Molecular Spectroscopy. 1993, 49, 2007‑2026.
CrossRef - Reed, A. E.; Curtiss, L. A.; Weinhold, F.Chem. Rev. 1988, 88, 899–926.
CrossRef - Reed, A. E.; Weinstock, R. B.; Weinhold,F. The Journal of Chemical Physics. 1985, 83,735‑746.
CrossRef - Domingo, L. R.; Chamorro, E.; Pérez, P. J. Org. Chem. 2008, 73, 4615–4624
CrossRef - Domingo, L. R.; Picher, M. T.Tetrahedron. 2004, 60, 5053‑5058.
CrossRef - Morell, C.; Grand, A.; Toro-Labbé, A. Chemical Physics Letters. 2006, 425,342‑346.
CrossRef - Morell, C.; Grand, A.; Toro-Labbé, A. J. Phys. Chem. A. 2005, 109,205‑212.
CrossRef - Domingo, L. R.RSC Adv.2014, 4, 32415‑32428.
CrossRef - Domingo, L. R.Molecules. 2016, 21, 1319-1334.
CrossRef

This work is licensed under a Creative Commons Attribution 4.0 International License.
![]()
A New Edition of Web of Science
Journal Impact Factor
2022: 0.5
Five Year: 0.8
Journal is Indexed in

Cabells Whitelist
![]()


