Vibrational Analysis and Non Linear Optical Activity of 3-fluoro-4–methylbenzonitrile
N. Y. Sugirtha Suni1, L. Guru Prasad2 and R. Ganapathi Raman1
1Department of Physics,1Nano Computational Laboratory, Department of Nano Technology, Noorul Islam Centre for Higher Education, Kumaracoil -629180, Thuckalay, India.
2Department of Science and Humanities, M.Kumarasamy College of Engineering, Karur.
Corresponding Author E-mail: ganapathiraman83@gmail.com
DOI : http://dx.doi.org/10.13005/ojc/340359
Article Received on : July 23, 2017
Article Accepted on : March 9, 2018
Article Published : 08 Jul 2018
The optimized molecular geometry, mulliken atomic charges, highest occupied molecular orbitals (HOMO) energy, lowest unoccupied molecular orbitals (LUMO) energy, polarizability and the first order hyperpolarizability of 3-fluoro-4-methylbenzonitrile has predicted with the help of quantum chemistry calculations by density functional theory (DFT) with B3LYP using 6-311++G(d,p) basis set. FTIR and FT-Raman spectra are investigated and compared with the observed data. Observed HOMO-LUMO energy gap offers the evidence for the presence of intermolecular interactions in the compound. First order hyperpolarizability calculated by quantum calculations infers that the title compound was an efficient tool for future applications in the field of non-linear optics. Natural bond orbitals and the thermodynamical properties were also studied by DFT.
KEYWORDS:3-Fluoro-4-Methylbenzonitrile; Density Functional Theory (DFT); FTIR; FT-Raman; HOMO; LUMO
Download this article as:| Copy the following to cite this article: Suni N. Y. S, Prasad L. G, Raman R. G. Vibrational Analysis and Non Linear Optical Activity of 3-fluoro-4–methylbenzonitrile. Orient J Chem 2018;34(3). |
| Copy the following to cite this URL: Suni N. Y. S, Prasad L. G, Raman R. G. Vibrational Analysis and Non Linear Optical Activity of 3-fluoro-4–methylbenzonitrile. Orient J Chem 2018;34(3). Available from: http://www.orientjchem.org/?p=45587 |
Introduction
Benzonitrile is an aromatic organic compound. Derivatives of benzonitrile find application in industries and medical field (M. Alcolea Palafox, 2003). Benzonitrile compounds are used as preservatives for food products. They are used for making aniline blue a dye. In medical field many benzonitrile derivatives in solid form are used as urinary antiseptic and vapour form are used for disinfecting bronchial tubes (Hermann Imgartinger, 2000). Since the derivatives of benzonitrile have wide applications,many studies are reported on such compounds. First order hyperpolarizability and HOMO-LUMO energy are the most important tools to predict the NLO activity of a compound. Quantum chemistry calculations provides the entire information about the structural, vibrational, electronic, optical, thermodynamic and other related properties of a molecule (David Pegu, 2013). Hence the present analysis was carried out to study the molecular properties of 3-fluoro- 4 – methylbenzonitrile and to elucidate useful information about the molecule.
Experimental Details
The compound 3-Fluoro-4-methylbenzonitrile (3F4MBN) was purchased from sigma-Aldrich Chemical Company, USA with a purity of not less than 99% and used as such for experimental studies. FT-Raman spectra of 3F4MBN was inscribed using 1064 nm line of Nd:YAG laser as the exciting wavelength in range 50-3500 cm-1 on a EZRaman, Enwaveoptronics, USA IFS 66 V spectrometer. Fourier transform infrared (FTIR) spectra was inscribed using 8400S Bruker, AlphaT,Germany infrared spectrophotometer using KBr pellet technique in the range 4000–400 cm-1. The spectra has been inscribed at normal temperature with a scanning speed 30 cm-1 min-1.
Computational Details
All calculations has been met with Gaussian 09 program package [M.J. Frisch, 2009] with the aid of DFT with B3LYP using 6-311++G(d,p) basis set and results were viewed using GAUSS VIEW program. HOMO and LUMO energy was obtained from time dependent density functional theory. NBO analysis has been executed using same basis set to study molecular interaction between filled and vacant orbitals. Polarizability and hyperpolarizability were also calculated.
Table 1: Geometrical Parameters of 3–fluoro- 4-methylbenzonitrile
| Parameter | B3LYP | Parameter | B3LYP | Parameter | B3LYP |
| BondLength(Å) | 6-311G++(d,p) | Bond Angle(°) | 6-311G++(d,p) | Dihedral angle | 6-311G++(d,p) |
| C1-C2 | 1.554 | C2-C1-H11 | 109.5741 | H11-C1-C2-C3 | -150.8946 |
| C1-H11 | 1.0675 | C2-C1-H12 | 108.9205 | H11-C1-C2-C7 | 29.386 |
| C1-H12 | 1.072 | C2-C1-H13 | 109.8545 | H12-C1-C2-C3 | 89.2887 |
| C1-H13 | 1.0814 | H11-C1-H12 | 109.5449 | H12-C1-C2-C7 | -90.4308 |
| C2-C3 | 1.3571 | H11-C1-H13 | 109.3711 | H13-C1-C2-C3 | -30.7176 |
| C2-C7 | 1.5456 | H12-C1-H13 | 109.562 | H13-C1-C2-C7 | 149.563 |
| C3-C4 | 1.5425 | C1-C2-C3 | 120.0591 | C1-C2-C3-C4 | -178.7972 |
| C3-H14 | 1.0713 | C1-C2-C7 | 119.9809 | C1-C2-C3-H14 | 1.1997 |
| C4-C5 | 1.3563 | C3-C2-C7 | 119.9594 | C7-C2-C3-C4 | 0.9223 |
| C4-H15 | 1.0712 | C2-C3-C4 | 119.8227 | C7-C2-C3-H14 | -179.0808 |
| C5-C6 | 1.5359 | C2-C3-H14 | 120.3491 | C1-C2-C7-C6 | 177.7006 |
| C5-C9 | 1.5345 | C4-C3-H14 | 119.8282 | C1-C2-C7-C8 | -2.5711 |
| C6-C7 | 1.362 | C3-C4-C5 | 120.2109 | C3-C2-C7-C6 | -2.0191 |
| C6-H16 | 1.0687 | C3-C4-H15 | 119.6262 | C3-C2-C7-C8 | 177.7092 |
| C7-F8 | 1.3528 | C5-C4-H15 | 120.1614 | C2-C3-C4-C5 | 1.422 |
| C9-N10 | 1.1563 | C4-C5-C6 | 120.064 | C2-C3-C4-H15 | -179.0196 |
| C4-C5-C9 | 119.8784 | H14-C3-C4-C5 | -178.5748 | ||
| C6-C5-C9 | 120.0563 | H14-C3-C4-H15 | 0.9835 | ||
| C5-C6-C7 | 119.9799 | C3-C4-C5-C6 | -2.6347 | ||
| C5-C6-H16 | 119.9932 | C3-C4-C5-C9 | 176.9544 | ||
| C7-C6-H16 | 120.0269 | H15-C4-C5-C6 | 177.8093 | ||
| C2-C7-C6 | 119.9001 | H15-C4-C5-C9 | -2.6016 | ||
| C2-C7-F8 | 120.2341 | C4-C5-C6-C7 | 1.5281 | ||
| C6-C7-C8 | 119.8652 | C4-C5-C6-H16 | -178.4505 | ||
| C9-C5-C6-C7 | -178.0602 | ||||
| C9-C5-C6-H16 | 1.9612 | ||||
| C5-C6-C7-C2 | 0.8132 | ||||
| C5-C6-C7-C8 | -178.9161 | ||||
| H16-C6-C7-C2 | -179.2082 |
Results and Discussion
Geometric Structure
The optimized geometrical structure of 3-fluoro- 4 – methylbenzonitrile is shown in Fig.1. The optimized bond length, bond angle and dihedral angle are calculated using B3LYP 6-311++G(d,p) basis set. The geometrical parameters calculated are shown in Table 1. These parameters can be utilized to elucidate other parameters of the compound under investigation.
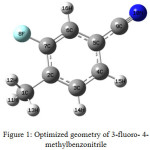 |
Figure 1: Optimized geometry of 3-fluoro- 4-methylbenzonitrile
|
Vibrational Analysis
The investigated compound has 16 atoms and so it possess 42 normal modes of vibrations. Vibrational frequencies calculated and observed are shown in table 2.
C-H vibrations
C-H stretching vibrations in aromatic compounds appear in the range 3100-3000 cm-1(M. Silverstein, 1989). In this study the peak at 3078 cm-1 and 3068 cm-1 in the FTIR spectrum and FT-Raman Spectra respectively are ascribed to C-H stretching vibrations. The corresponding calculated values are 3086 cm-1and 3060 cm-1which are in accordance with the observed values. For substituted benzenes, the three in-plane C-H vibrations appear in a range 1300-1000 cm-1and three out-of-plane bending vibrations appear in a range 1000-750 cm-1(J. Sharmi Kumar, 2015). The peaks at 1142, 1194, 1214 , 1252 cm-1 and at 1132, 1200 cm-1 in the FTIR spectrum and FT-Raman Spectrum are ascribed to in-plane C-H bending vibrations which are in accordance with the calculated values 1130, 1171, 1216 and 1276 cm-1 . The peaks at 831, 886 and 951 cm-1 and at 867, 936 cm-1 in the FTIR spectrum in the FT-Raman Spectrum are ascribed to out- of -plane C-H bending vibrations which are in accordance with the calculated values 836, 896 and 950 cm-1.
C-C vibrations: Ring C-C stretching vibration appears in a range 1650-1400 cm-1(N. Sundaraganasan, 2009). Peaks at 1492 cm-1, 1562 cm-1 and at 1494 cm-1, 1591 cm-1 in the FTIR spectrum and FT-Raman Spectrum are ascribed to C-C vibrations. The corresponding worked out values are 1494 cm-1 , 1564 cm-1 and 1492 cm-1 ,1531 cm-1 which are in accordance with the observed data.
C-F vibrations: C-F vibration appears in the range 1360-1000 cm-1 (K. Sambathkumar, 2015). The sharp peak at 1270 cm-1 and at 1285 cm-1 in the FTIR spectrum and FT-Raman Spectra are ascribed to C-F vibrations and are in accordance with calculated values 1270 cm-1and 1276 cm-1.
Vibrations: vibration appears around 2200 cm-1 (S.Gunasekaran, 2006). The peak at 2244 cm-1 and at 2221 cm-1 in the FTIR spectrum and FT-Raman Spectra are ascribed to vibrations which are in accordance with calculated values 2242 cm-1and 2238 cm-1.
CH3group vibrations: The title compound has only one substituted methyl (CH3) group in the fourth position of the benzene ring. A methyl group is associated with nine fundamental mode of vibrations namely , the symmetric stretching mode (CH3 sym. stretch) , asymmetric stretching mode (CH3 asym. stretch), in-plane hydrogen stretching mode, the symmetric deformation mode(CH3 sym. deform) , asymmetric deformation mode(CH3 asy. deform), the in-plane rocking mode (CH3 ipr), out-of-plane rocking mode (CH3 opr) and twisting (tCH3) mode. Substituted methyl groups in the aromatic ring systems are typically specified as electron donating groups (D. Lin-Vein, 1991).
Generally CH3 vibration appear in a range (2900-3000 cm-1)(M. Murugan, 2012). Peak at 3000 cm-1 and at 2986 cm-1 in the FTIR spectrum and FT-Raman Spectra are ascribed to CH3 symmetrical stretching mode vibration which are in accordance with the calculated value 3035cm-1. The peak at 1499cm-1 in the FTIR spectrum corresponds to CH3 in plane bending modes which is in accordance with the calculated value 1492cm-1. The peak at 1069cm-1 in the FTIR spectra harmonize to CH3 in rocking mode vibration which is in accordance with the calculated value 1060cm-1.
Table 2: Observed and calculated (FT-IR, FT-Raman) vibrational frequencies of the title compound.
| Mode | Label | Experimental(cm-1)FT-IR | FT-Raman | B3LYP/6-311++G(d,p) | IR Intensity(Km/mol) | Raman Activity | Vibrational assignments |
| 1 | A | 95.7103 | 0.0009 | 0.3696 | τCH3 | ||
| 2 | A | 104.1663 | 1.438 | 0.4454 | τC≡N | ||
| 3 | A | 154 | 148.0831 | 5.0946 | 3.2263 | βC≡N | |
| 4 | A | 194 | 197.8587 | 3.2846 | 1.3948 | γC-CH3+γC≡N | |
| 5 | A | 273.7 | 276.2751 | 2.2767 | 0.3873 | βC-CH3+βC-F | |
| 6 | A | 280 | 282.9399 | 2.3928 | 0.6734 | ω C-F | |
| 7 | A | 414 | 409.8928 | 1.4935 | 4.0477 | γC-CH3 | |
| 8 | A | 423 | 432.2994 | 1.4908 | 3.9706 | 16a γ C-C-C | |
| 9 | A | 434 | 436.6913 | 1.945 | 1.5209 | β C-C-C | |
| 10 | A | 513.3 | 462.6 | 491.5191 | 2.5994 | 1.6762 | 16b γ C-C-C |
| 11 | A | 560.4 | 544 | 545.9524 | 3.7765 | 7.3407 | 16b β C-C-C |
| 12 | A | 611.8 | 615.8 | 603.8151 | 2.6259 | 0.9818 | 16a β C-C-C |
| 13 | A | 632.4 | 620 | 643.1389 | 10.0649 | 1.2955 | 6a δ |
| 14 | A | 685.8 | 700.6038 | 0.7185 | 2.4935 | γ C-C-C+t C≡N | |
| 15 | A | 753.2 | 754 | 713.601 | 1.7294 | 0.3961 | γ C-C-C+t C≡N |
| 16 | A | 768.3 | 769.1046 | 5.0664 | 24.505 | β C-C-C+t C-F | |
| 17 | A | 831.4 | 836.704 | 23.2161 | 0.1268 | 17b γ C-H | |
| 18 | A | 886.4 | 867 | 896.4401 | 24.4011 | 0.1313 | γ C-H+τ C≡N |
| 19 | A | 951.45 | 936 | 950.8395 | 21.1946 | 4.5349 | 10a γ C-H |
| 20 | A | 993.8 | 970.5941 | 0.0134 | 0.0512 | Ring breathing | |
| 21 | A | 1007.4 | 1006 | 1019.558 | 18.0624 | 0.9225 | Trigonal bending |
| 22 | A | 1069 | 1060.461 | 2.6859 | 0.0411 | ρ CH3 | |
| 23 | A | 1142.5 | 1132 | 1130.859 | 36.0379 | 19.7862 | β CH+ υ C-F |
| 24 | A | 1194.9 | 1171.555 | 0.575 | 3.0025 | β CH | |
| 25 | A | 1214.5 | 1200 | 1216.88 | 3.7437 | 1.9298 | β CH+ υ C-F |
| 26 | A | 1270.4 | 1277 | 1276.279 | 78.5627 | 76.1255 | β CH |
| 27 | A | 1285 | 1295.275 | 1.0495 | 1.5283 | υ C-F | |
| 28 | A | 1332 | 1329.431 | 0.409 | 2.3016 | γC-CH3 | |
| 29 | A | 1429.5 | 1410 | 1419.072 | 3.2555 | 20.0889 | CH3 asym.deform |
| 30 | A | 1469.5 | 1439.191 | 29.835 | 0.7039 | CH3 asym.deform | |
| 31 | A | 1472 | 1487 | 1480.588 | 8.2457 | 9.5451 | 14 υ C-C |
| 32 | A | 1499 | 1492.38 | 8.7334 | 13.1925 | β CH3 | |
| 33 | A | 1572.6 | 1584.7 | 1531.799 | 50.2532 | 1.5091 | 19a υ C-C |
| 34 | A | 1600 | 1601.005 | 40.2309 | 2.2114 | 8b υ C-C (semi-circle stretch) | |
| 35 | A | 1668.6 | 1658 | 1656.248 | 2.154 | 172.8537 | 8a υ C-C |
| 36 | A | 2338 | 2338 | 2333.982 | 43.5827 | 557.747 | γ C≡N |
| 37 | A | 3000 | 2986 | 3035.594 | 15.3871 | 306.9231 | υsym CH3 |
| 38 | A | 3078 | 3053 | 3086.357 | 8.4141 | 99.1312 | 20a** arom. υ C-H |
| 39 | A | 3117 | 3108 | 3118.279 | 12.3515 | 65.7333 | υ C-H |
| 40 | A | 3178 | 3177.419 | 5.5481 | 75.475 | υ C-H | |
| 41 | A | 3203 | 3207 | 3203.313 | 1.539 | 102.9211 | υ C-H |
| 42 | A | 3221 | 3212.26 | 0.7115 | 88.4629 | υ C-H |
υ-stretching; υsym-symmetrical stretching; υasy– asymmetrical stretching; β-in plane bending; γ-out-of-plane bending; ω-wagging; t-twisting; δ-scissoring; τ-torsion;ρ-rocking; *-wilson’s notion; IR int-IR intensities.
Mulliken Atomic Charges
The scope of bonding of a molecule depend on the number of unpaired electrons in the atoms and hence the atomic charges has been retrieved by Mulliken population assay (A.A. Popov, 2004). Mulliken atomic Charges calculation plays an important part in applying quantum chemistry calculation to molecular systems because atomic charge affects dipole moments, polarizability, electronic structures and other properties of molecular systems (R.S.Mulliken, 1985). Mulliken charges obtained using B3LYP 6-311++G (d,p) are shown in Table 3. Mulliken atomic charges graph is shown in Fig.2. All hydrogen atoms exhibits positive charge, nitrogen and fluorine atom exhibit negative charge .This suggests the creation of intermolecular interaction in solid forms(Isa Sidir, 2010).From the charge calculation it is clear that nitrogen atom having negative charge acts as donor atom and the ring hydrogen atom having positive charge acts as acceptor atoms.
Table 3: MullikenPopulationAnalysis of 3–fluoro- 4-methylbenzonitrile
| Sl.No | Atoms | B3LYP |
| 1 | C | -0.38354 |
| 2 | C | 1.303938 |
| 3 | C | -0.85876 |
| 4 | C | -0.53413 |
| 5 | C | 2.179886 |
| 6 | C | -0.15444 |
| 7 | C | -0.68165 |
| 8 | F | -0.17206 |
| 9 | C | -1.58496 |
| 10 | N | -0.1711 |
| 11 | H | 0.178828 |
| 12 | H | 0.17881 |
| 13 | H | 0.157086 |
| 14 | H | 0.143043 |
| 15 | H | 0.190491 |
| 16 | H | 0.208561 |
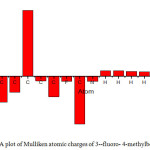 |
Figure 2: A plot of Mulliken atomic charges of 3–fluoro- 4-methylbenzonitrile |
Polarizability and Hyperpolarizability
The reaction of systems in applied electric fields has been explained by its Polarizability and hyperpolarizability. The non linear optical property of a compound can be studied using these parameters. The investigated first order hyperpolarizability of investigated compound is 2.768×10-30 esu which is 9 times urea (0.2991×10-30esu) a standard NLO material (Li Xiao-Hong,2011). Calculated dipole moment, polarizability and hyperpolarizability are given in Table 4.
Table 4: Electric dipole moment, polarizability and hyperpolarizability of 3–fluoro- 4-methylbenzonitrile.
| Parameters | B3LYP |
| 6-311++G(d,p) | |
| μ x | 0.5802156 |
| μ y | 0.010185 |
| μ z | -1.7797845 |
| μ= | 1.87200Debye |
| α xx | 92.0760016 |
| α xy | -0.0391977 |
| α yy | 54.1494 |
| α xz | 0.300154 |
| α yz | -0.5182233 |
| α zz | 145.5895464 |
| α0 | 194.7552×10-33esu |
| α= | 1682.549×10-33esu |
| β xxx | 103.2968388 |
| β xxy | -7.7277807 |
| β xyy | 34.3821757 |
| β yyy | 6.763047 |
| β xxz | 24.3731807 |
| β xyz | 0.135037 |
| β yyz | -18.6541887 |
| β xzz | 28.6181066 |
| β yzz | 5.1407 |
| β zzz | 268.1870903 |
| β0 | 2768.5784×10-33esu |
HOMO LUMO Analysis
HOMO stands for highest occupied molecular orbital which represents the ability of a molecule to donate an electron and LUMO stands for lowest unoccupied molecular orbital which represents the ability of a molecule to accept an electron. HOMO and LUMO are the major orbitals that take part in the chemical stability of the molecule (J.A. Alanso,2004). The calculated HOMO LUMO gap using B3LYP 6-311++G(d,p) is 5.61eV. The HOMO LUMO energy gap explain that the title compound is experiencing charge transfer interactions and it reflects its NLO property (Basak Kosar,2011). The calculated energy values are shown in Table 5.
Table 5: Calculated energies of 3–fluoro- 4-methylbenzonitrile.
| LUMO | -1.79 |
| HOMO | -7.4 |
| Energy Gap | 5.61 |
| Electronegativity (χ) | -4.595 |
| Chemical Potential (µ) | 4.595 |
| Global Hardness (ƞ) | 2.805 |
| Global softness (s) | 0.356506239 |
| Electrophilicity Index (ω) | 6.4444875 |
| EHOMO-1(eV) | -7.82 |
| ELUMO+1(eV) | -1.36 |
| EHOMO-1 – ELUMO+1(eV) | -6.46 |
Thermodynamic Parameters
Several thermodynamical parameters has been calculated and are listed in Table 6. Scale factors were recommended (Zeynep Demircioglu, 2014) for calculating zero point vibrational energy and entropy accurately. Changes in total energy and entropy at normal temperature are presented in Table.6. These changes seems to be insignificant.
Table 6: Thermodynamic parameters
| Thermodynamic functions Of DMAP | B3LYP |
| 6-311++G(d,p) | |
| Self-consistent field energy (a.u) | -463.132 |
| Zero point vibrational energy (kcal/mol) | 74.043 |
| Rotational constant (GHz) | 3.021 |
| 0.880 | |
| 0.684 | |
| Rotational temperature (K) | 0.145 |
| 0.042 | |
| 0.033 | |
| Thermal energy (kcal/mol) | |
| Total | 79.491 |
| Translational | 0.889 |
| Rotational | 0.889 |
| Vibrational | 77.714 |
| Specific heat capacity at constant volume (cal/mol K) | |
| Total | 32.233 |
| Translational | 2.981 |
| Rotational | 2.981 |
| Vibrational | 26.271 |
| Dipole moment (Debye) | 4.7644 |
| Lumo(eV) | -1.79 |
| Homo(eV) | -7.4 |
| Energy gap(eV) | -5.61 |
| Entropy(S)(cal/mol K) | |
| Total | 91.514 |
| Translational | 40.614 |
| Rotational | 29.559 |
| Vibrational | 21.342 |
| Gibbs Free Energy | 0.084 |
| Enthalpy | 0.128 |
Non Linear Optical Activity
NLO activity give key function for properties like the frequency shifting, optical modulation, optical swaping , optical logic for the extending technology in the field of communications, signal processings and optical inter-connections(Mauricio Alcolea Palafox, 2000).Molecules that exhibit asymmetric polarization which is induced because of electron donars and acceptors in the pi-electron conjugatedsystems are efficient materials for electro-optics and NLO applications (I.Khan, 2013). In order to find the non linear activity of the material, first order hyperpolarizability of the investigated compound was calculated and compared with urea, a standard NLO material. It was found that the first order hyperpolarizability of our investigated compound is 9 times than urea. Hence we propose that the investigated compound under study is an efficient material for future NLO applications.
Conclusion
Detailed investigation of the structural and electronic property of the compound under study has been performed by DFT using suitable basis set. Calculated first order hyperpolarizability and HOMO- LUMO energy gap confirmed the NLO property of the compound. First order hyperpolarizability calculated for the compound is 9 times greater than urea. Hence the compound under study is an efficient material for future NLO applications. Mulliken atomic charge calculation suggests that there is charge transfer from N, F to H.
References
- M. Alcolea Palafox, V. K. Rastogi, L. Mittal, “Benzonitriles: Survey of their importance and scaling of their vibrational frequencies,” Int.J.Quantum Chem., vol. 94, pp 189-204, January 2003.
CrossRef - Hermann Imgartinger, Peter Walter Fettel, Thomas Escher , Philip Tinnefeld, Simon Nord, and Markus Saucer “Substituent effects on Redox properties and photoinduced electron transfer in Isoxazolo Fullerenes,” Eur. J.Org.Chem., vol.2000, pp 455-465, January 2000.
CrossRef - David Pegu and Ngangbam Bedamani Singh, “Quantum Chemical Calculations of Molecular Structure, Electronic, Thermodynamic and Non-linear optical properties of 2-amino-3-nitro-6-methylpyridine,”Int.J.Advanced Research, vol.1, issue 9, pp 531-538, November 2013.
- M. J. Frisch, et al., GAUSSIAN 09, Revision A. 9, Gaussian, INC, Pittsburgh, 2009.
- M. Silverstein, G.C. Basseler, C. Morill, Spectrometric Identification of Organic Compounds, Wiley, New York, 1981.
- J. Sharmi Kumar, T. S. Renuga Devi, G. R. Ram Kumaar, A.Bright, “Ab initio and density functional theory calculations of molecular structure and vibrational spectra of 4-(2-Hydroxyethyl) piperazine-1-ethanesulfonic acid,” Spectrochimica Acta Part A: Molecular and Biomolecular Spectroscopy, vol.152, pp 509-522, July 2015.
CrossRef - N. Sundaraganasan, G. Elango, S. Sebastian, & P. Subramani, Ind. J. Pure App. Phy. 47 (2009) 481.
- K. Sambathkumar, S. Jeyavijayan, M. Arivazhagan, Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 147 (2015) 124.
CrossRef - S.Gunasekaran, S.Seshadri, S.Muthu, Indian J.Pure and Applied Physics, vol.44, pp360-366, 2006.
- D. Lin-Vein, N.B. Colthup, W.G.Fateley, J.G.Grasselli, “The Handbook of Infrared and Raman Characteristics Frequencies of Organic Molecules, Academic Press, San Diego, CA, 1991.
- M. Murugan, V.Balachandran and Marana, “Vibrational spectra and electrostatic potential surface of 2-fluoro-6-methoxybenzonitrile based on quantum chemical calculations,” J. Chemical and Pharmaceutical Research, vol.4, issue 7, pp 3400-3413, 2012.
- A.A. Popov, V. M. Senyavin, A.A. Ganovsky, Chem.Phys.Lett, 383 (2004) 149-155.
CrossRef - R. S. Mulliken, J. chem. phys., vol 23, pp 1833-1840, 1985.
CrossRef - Isa Sidir, Yadigar Gulseven Sidir, Mustafa Kumalar, Erol Tasal, “Ab initio Hartree-Fock and density functional theory investigations on the conformational stability, molecular structure and vibrational spectra of 7-acetoxy-6-(2, 3-dibromopropyl)-4, 8-dimethyl conmarin molecule,” J. Mol. Struct. Vol.964 pp 134-151, February 2010.
CrossRef - Li Xiao-Hong, Liu Xiang-Ru, Zhang Xian-Zhou, “Calculation of vibrational spectroscopic and NMR parameters of 2- Dicyanovinyl-5-(4-N, N- dimethyl aminophenyl) thiophene by ab initio HF and density functional methods,” Comput.Theor.Chem. vol. 969, pp 27-34, August 2011.
CrossRef - J.A. Alanso , L.C. Balbas and A.Rubio, “ Non local functional for exchange and correlation in density functional theory Application to atoms and to small atomic clusters,” Int. J. Quantum Chem. Vol. 56 pp 499-508, September 2004.
CrossRef - Basak Kosar, C. Albayrak, “Spectroscopic investigations and quantum chemical compound study of (E) -4-methoxy-2-[p-tolylimino) methyl] phenol,”Spectrochim Acta,vol. 78A, pp 160-167, January 2011.
CrossRef - Zeynep Demircioglu, Cigdem Albayrak, Orhan Buyukgungor, “Experimental (X-ray, FT-IR and UV-Vis spectra) and theoretical methods (DFT study) of (E)-3 –methoxy -2-[p- tolylimino) methyl ] phenol,” Spectrochim Acta, vol. 128A, pp 748-758, July 2014.
CrossRef - Mauricio Alcolea Pala fox,“Scaling factors for the prediction of vibrational spectra. I. Benzen molecule,” Int. J. Quantum Chem. Vol. 77,pp 661-684, March 2000.
CrossRef - I. Khan, A.Ahmad, J.Phys.chem.solid 74 (2013) 1818.
CrossRef

This work is licensed under a Creative Commons Attribution 4.0 International License.
![]()
A New Edition of Web of Science
Journal Impact Factor
2022: 0.5
Five Year: 0.8
Journal is Indexed in

Cabells Whitelist
![]()


