Molecular Docking as A Computational Tool for Analyzing Product Mediated Inhibition for β-Galactosidase Immobilized on Glutaraldehyde Modified Matrices
Shakeel Ahmed Ansari1, Mohammad Alam Jafri1, Rukhsana Satar2, Syed Ismail Ahmad3 and Sandesh Chibber4
1Center of Excellence in Genomic Medicine Research, King Abdulaziz University, Jeddah-21589, Kingdom of Saudi Arabia.
2Department of Biochemistry, Ibn Sina National College for Medical Studies, Jeddah-21418, Kingdom of Saudi Arabia.
3Physics, Department of Basic Sciences, Ibn Sina National College for Medical Studies, Jeddah-21418, Kingdom of Saudi Arabia.
4Biological and Life Sciences, School of Arts and Sciences, Ahmedabad University, Ahmedabad 380009, India.
Corresponding Author E-mail: sandesh.chibber@ils.ahduni.edu.in
DOI : http://dx.doi.org/10.13005/ojc/340227
Article Received on : September 18, 2017
Article Accepted on : December 30, 2017
Chemical informatics aims to disseminate information regarding the design and structure of a compound for revealing chemical information of a target with the help of molecular modeling/simulation. Hence, in the present study, efforts were raised to analyze the ligand interaction plots of glucose and galactose for Aspergillus oryzae β-galactosidase. The crystallographic structure of enzyme was obtained from protein data bank ID: 4IUG while the 3D structure of glutaraldehyde was obtained from PubChem with compound ID 3485. It was prepared by Chimera v.1.6.2 for hydrogen and charge addition. Chimera v.1.6.2 and PyMOL v.1.3 were used for visual analyses and illustration of protein-ligand complex. The ligand interaction plots of protein-ligand complexes were illustrated by Ligplot+ v.1.4.5 program. Enzyme showed optimum binding affinity on a molecular target with the binding energy of −9.5 kcal/mol.
KEYWORDS:β-galactosidase; Chimera v.1.6.2; Galactose; In Silico Analysis; PyMOL v.1.3
Download this article as:| Copy the following to cite this article: Ansari S. A, Jafri M. A, Satar R, Ahmad S. I, Chibber S. Molecular Docking as A Computational Tool for Analyzing Product Mediated Inhibition for β-Galactosidase Immobilized on Glutaraldehyde Modified Matrices. Orient J Chem 2018;34(2). |
| Copy the following to cite this URL: Ansari S. A, Jafri M. A, Satar R, Ahmad S. I, Chibber S. Molecular Docking as A Computational Tool for Analyzing Product Mediated Inhibition for β-Galactosidase Immobilized on Glutaraldehyde Modified Matrices. Orient J Chem 2018;34(2). Available from: http://www.orientjchem.org/?p=43880 |
Introduction
β-galactosidase (EC.3.2.1.23) is a glycosidic hydrolase that catalyzes the hydrolysis of β-galactosides into monosaccharides by breaking glycosidic bonds [1]. Because of low levels of this enzyme in intestine, large fraction of the population shows lactose intolerance and they have difficulty in consuming dairy products. Hence, treatment of such products with lactase serves as an appropriate method to reduce their lactose content for lactose intolerant population [2, 3]. Moreover, the hydrolysis of whey converts lactose into a very useful product like sweet syrup, which can be used in various processes of dairy, confectionary, baking and soft drink industries. Therefore, lactose hydrolysis not only allows the milk consumption by lactose intolerant population but can also solve the environmental problems linked with whey disposal [4].
Product inhibition by galactose and glucose often causes a serious loss in productivity of soluble β-galactosidase for lactose conversion. These drawbacks are overcome by the immobilization of enzyme and glutaraldehyde has been used in the recent past either as crosslinking agent or for modifying the surface of matrix for improving the stability of enzyme [5-11].
Protein engineering strategies are developed for constructing enzymes with novel/improved activities, specificities and stabilities via in-silico methods. Computational methods can be principally grouped into three main categories: bioinformatics, molecular modeling and de-novo design [12, 13]. The de-novo protein design is experiencing rapid development thereby resulting in more robust and reliable predictions [14]. It might therefore be possible to deploy algorithms that identify protein sequences folding into a known 3D structure. In this regard, molecular docking is a frequently used method for structure-based rational drug design [15]. It is used for evaluating the complex formation of small ligands with large biomolecules, predicting the strength of the bonding forces and finding the best geometrical arrangements to know about the concepts of protein structure, enzyme-inhibitor interactions, intermolecular forces and role of molecular design in drug-development [Fig 1].
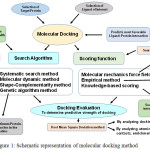 |
Figure 1: Schematic representation of molecular docking method Click here to View figure |
Molecular docking is a computational technique which involves various search and scoring methods to ascertain most favorable binding affinity between a protein and selected ligand.
Computer-based methods are becoming increasingly important apart from wet laboratory experiments for studying the structure and function of biomolecules. Although early efforts were hindered by limited possibilities in computational resources, however, due to recent advances in high performance computing softwares, virtual screening methods became more and more efficient. These methods contributed to the development of highly efficient protein-ligand complex in biotechnology industries [16, 17]. Examples include molecular docking studies of the inhibitions of angiotensin converting enzyme and renin activities by hemp seed peptides [18] and quercetin glycosides [19]. Similarly, structural and functional studies of β-galactosidase have been studied by interaction with sodium dodecyl sulfate [20] and ethanol functionalized graphene [1] for elucidating their efficacy in biotechnology industries. Docking programs simulate how a target macromolecule (receptor, enzyme or nucleic acid) interacts with small molecule ligands such as substrate, inhibitor or other drug candidate. To model the binding between the ligand and the target molecule, their known 3D structures are superimposed and fit between the key sites of target molecule, and ligand is then analyzed. By using molecular mechanics, the programs usually determine the binding energy between the hosts’s binding site and ligand, a feature used to predict and describe the efficacy of the binding.
In this study, automated active site identification, docking and scoring protocol for galactose and glucose on β-galactosidase targets based on physicochemical descriptors has been performed, to reveal the fact that galactose (competitive inhibitor) can enter substrate binding region of enzyme active site.
Methodology
Accession of Target Protein
Three-dimensional structure of β-galactosidase (from Aspergillus oryzae) in complex with galactose was obtained from Protein Data Bank with ID: 4IUG.
Ligand Selection
Three-dimensional structure of glutaraldehyde was obtained from PubChem (https://pubchem.ncbi.nlm.nih.gov/) with Compound ID 3485. It was checked/prepared by Chimera v.1.6.2 for hydrogen and charge addition [21]. PubChem ID of D-glucose was CID: 5793.
Target and Ligand Optimization
Initial structural preparation of β-galactosidase which was required for docking was analyzed by Dock v.6.5 and performed by Chimera v.1.6.2 [21]. The rigid body docking option of Dock v.6.5 [22] was used with default parameters. Chimera v.1.6.2 [Pettersen et al., 2004] and PyMOL v.1.3 [23] were used for visual analyses and illustration of protein-ligand complex. The ligand interaction plots of protein-ligand complexes were illustrated by Ligplot+ v.1.4.5 program [24, 25]. At each step of the docking simulation, interaction energy of ligand and protein was evaluated using atomic affinity potentials computed on a grid. The remaining parameters were set as default.
Results and Discussion
Since protein-ligand complexes are rather difficult to study using experimental tools, computational approaches had shown some promise in this regard [26]. Thus, docking was used as an important computational tool for specific prediction of protein-ligand interactions by determining the accuracy and scoring reliability. As for the former, it indicates how similar the prediction of ligand binding is to the ligand conformation that is determined experimentally whereas scoring reliability ranks ligands based on their affinities [15, 17, 27, 28]. Following these type of studies, a data repository can be created and utilized by researchers for the submission and retrieval of information on β-galactosidases from various sources, in order to utilize them efficiently in biotechnology industries.
Glutaraldehyde is a bi-functional reagent with the capacity to polymerize. It reacts with different enzyme moieties via primary amino groups of proteins and other groups like thiols, phenols and imidazoles [29]. It is a widely accepted fact that glutaraldehyde imparts stability to β-galactosidase as a result of binding to immobilization matrix by forming strong bonds and acting as a crosslinking agent.
Fig 2 showed the dock/grid score (-22) obtained was negative with high absolute value. Seven interacting residues revealed from docking analyses were Tyr-96, Asn-140, Glu-142, Asn-199, Tyr-260, Phe-264 and Glu-298 which form non-bonding interactions (and H bond) with glutaraldehyde to make the complex highly stable. The hydrogen bond (3.10 Å) exists between NH2 carboxamide side chain of Asn-199 and “O” atom of one of the aldehyde group of glutaraldehyde. Fig 3 suggested that while comparing glutaraldehyde binding to that of natural substrate i.e. galactose, it was observed that five interacting residues Tyr-96, Asn-140, Glu-142, Tyr-260, and Glu-298 were overlapping. These findings explained the fact that glutaraldehyde binds to the similar region in the galactose binding site and interferes with the binding of galactose for showing competitive inhibition for β-galactosidase. Such stacking interactions can be used to rank the molecular docking and matching between inhibitors and enzymes. Considering the physical properties of these interacting molecules by molecular docking concepts, analysis can be done on membrane/matrix surface along the molecular dynamics run and the features of interface between membrane and membrane-binding molecule. It was also proposed earlier that angiotensin-converting enzyme (ACE) docked successfully with quercetin glycosides [19].
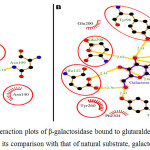 |
Figure 2: The ligand interaction plots of β-galactosidase bound to glutaraldehyde modified matrices (A) and its comparison with that of natural substrate, galactose (B) Click here to View figure |
The residues forming hydrophobic interactions are shown as red arcs while the hydrogen bonds are shown as yellow dashed lines with indicated bond lengths (in Angstrom). The interacting residues which are common for both the ligands are encircled.
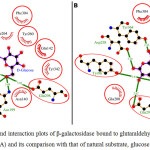 |
Figure 3: The ligand interaction plots of β-galactosidase bound to glutaraldehyde modified matrices (A) and its comparison with that of natural substrate, glucose (B) Click here to View figure |
The residues forming hydrophobic interactions are shown as red arcs while the hydrogen bonds are shown as yellow dashed lines with indicated bond lengths (in Angstrom). The interacting residues which are common for both the ligands are encircled.
These researchers observed that possible binding modes of quercetin at ACE active sites includes Arg 124, Tyr 135, Ile 204, Ala 208, Glu 216, Tyr 215, Glu 96 with quercetin ligand molecule. Moreover, docking score of -28.04 was obtained for D-Glucose. The probable reason is that glucose and galactose are epimers and the only difference between them is difference in orientation of -OH group at 4th carbon. The ligand showed significant interaction with target proteins based on Root Mean Square Deviation (RMSD) values as compared to standard. Beside RMSD clustering, AutoDock v.6.5 has also calculated the binding free energies of these interactive molecules to find the best binding mode. The calculated final docked energy for galactose was -9.5 kcal/mol. Hence, the docking results revealed that such compounds can enter the substrate-binding region of the enzyme active site.
Conclusion
The generation and visualization of enzyme-inhibitor binding data obtained by molecular docking provide opportunity to analyze the ligand interaction plots of glucose and galactose for the enzyme β-galactosidase. Such studies can lead to creation of data repository for the submission and retrieval of information on plethora of enzymes, in order to utilize them efficiently in biomedical and biotechnological applications.
Acknowledgment
The authors are thankful to the Center for providing all the necessary facilities to carry out this work.
References
- Satar, R.; Ismail, S.A.; Rehan.; M, Ansari, S.A. Bioproc. Biosyst. Eng. 2016, 39, 807-814.
CrossRef - Ansari, S.A.; Satar, R. J. Mol. Catal. B: Enzym. 2012, 81, 1-6.
CrossRef - Brown III, C.W.; Oh, E.; Hastman Jr, D.A.; Walper, S.A.; Susumu, K.; Stewart, M.; Deschamps,J.R.; Medintz, I.L. RSC Adv. 2015, 5, 93089-93094.
CrossRef - Benavente, R.; Pessela, B.C.; Curiel, J.A.; Rivas, B.; Munoz, R.; Guisan, J.M.; Mancheno, J.M.; Cardelle-Cobas, A.; Ruiz-Matute, A.I.; Corzo, N. Molecules 2015, 30, 7874-7889.
CrossRef - Ozdural, A.R.; Tanyolac, D.; Boyac, I.H.; Mutlu, M.; Webb, C. Biochem. Eng. J. 2003, 14, 27-36.
CrossRef - Ansari, S.A.; Satar, R.; Zaidi, S.K. Quim. Nov. 2015, 38, 387-392.
- Nguyen, T.H.; Splenchtna, B.; Steinbock, M.; Kneifel, W.; Lettner, H.P.; Kulbe, K.D.; Haltrich, D. J. Agric. Food Chem. 2006, 54, 4989-4998.
CrossRef - Hsu, C.A.; Lee, S.L.; Chou, C.C. J. Agric. Food Chem. 2007, 55, 2225-2230.
CrossRef - Park, A.; Oh, D. Appl. Microbiol. Biotechnol. 2010, 85, 1427-1435.
CrossRef - Pessela, B.C.C.; Mateo, C.; Carrascosa, A.V.; Vian, A.; Garcıa,, J.L.; Rivas, G.; Alfonso, C.; Guisan, J.M.; Fernandez-Lafuente, R. Biomacromolecules 2003, 4, 107-113.
CrossRef - Mateo, C.; Monti, R.; Pessela, B.C.C.; Fuentes, M.; Torres, R.; Guisan, J.M.; Fernandez-Lafuente, R. Biotechnol. Prog. 2004, 20, 1259-1262.
CrossRef - Damborsky, J.; Brezovsky, J. Curr. Opin. Chem. Biol. 2014, 19, 8-16
CrossRef - Dhanjal, J.K.; Sreenidhi, A.K.; Bafna, K.; Katiyar, S.P.; Goyal, S.; Grover, A.; Sundar, D. Plos One 2015, 10, e0134691.
CrossRef - Kries, H.; Blomberg, R.; Hilvert, D. Curr. Opin. Chem. Biol. 2013, 17, 221-228.
CrossRef - Singh, T.; Biswas, D.; Jayaram, B. J. Chem. Inf. Model. 2011, 51, 2515-2527.
CrossRef - Neves, M.A.C.; Totrov, M.; Abagyan, R. J. Comput. Aid. Mol. Des. 2012, 26, 675-686.
CrossRef - Sousa, S.F.; Ribeiro, A.J.M.; Coimbra, J.T.S.; Neves, R.P.S.; Martins, S.A.; Moorthy, N.S.H.N.; Fernandes, P.A.; Ramos, M.J. Curr. Med. Chem. 2013, 20, 2296-2314.
CrossRef - Girgih, A.T.; He, R.; Aluko, R.E. J. Agric. Food Chem. 2014, 62, 4135-4144.
CrossRef - Muhammad, S.A.; Fatima, N. Pharmacogn. Mag. 2015, 11, S123-S126.
CrossRef - Muga, A.; Arrondo, J.L.; Bellon, T.; Sancho, J.; Bernabeu, C. Arch. Biochem. Biophys. 1993, 300, 451-457.
CrossRef - Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. J. Comp. Chem. 2004, 25, 1605-1612.
CrossRef - Ewing, T.J.; Makino, S.; Skillman, A.G.; Kuntz, I.D. J. Comp. Aided Mol. Des. 2001, 15, 411-428.
CrossRef - Delano, W.L. San Carlos, CA. DeLano Scientific (2002).
- Wallace, A.C.; Laskowski, R.A.; Thornton, J.M. Prot. Eng. 1995, 8, 127-134.
CrossRef - Laskowski, R.A.; Swindells, M.B. J. Chem. Inf. Model. 2011, 51, 2778-2786.
CrossRef - Panneerselvam, S.; Yesudhas, D.; Durai, P.; Anwar, M.A.; Gosu, V.; Choi, S. Molecules 2015, 20, 14915-14935.
CrossRef - Rudnitskaya, A.; Torok, B.; Torok, M. Biochem. Mol. Biol. Educ. 2010, 38, 261-265.
CrossRef - Chibber, S.; Ahmad, I. Biochem. Biophys. Rep. 2016, 6, 63-67.
- Barbosa, O.; Ortiz, C.; Berenguer-Murcia, A.; Torres, R.; Rodrigues, R.C.; Fernandez-Lafuente, R. RSC Adv. 2014, 4, 1583-1590.
CrossRef

This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 4.0 International License.

This work is licensed under a Creative Commons Attribution 4.0 International License.
![]()
A New Edition of Web of Science
Journal Impact Factor
2022: 0.5
Five Year: 0.8
Journal is Indexed in

Cabells Whitelist
![]()


