Synthesis, Characterization and Cytotoxic Activity of Betulinic Acid and Seco-Betulinic Acid Derivatives against Human Colorectal Carcinoma
Mohd Tajudin Mohd Ali1, Habsah Zahari1, Aisyah Aliasak1, Siong Meng Lim2,3, Kalavathy Ramasamy2,3 and Allan Patrick G. Macabeo4
1Faculty of Applied Sciences, Universiti Teknologi MARA(UiTM),40450, Shah Alam, Selangor, Malaysia.
2Faculty of Pharmacy, Universiti Teknologi MARA (UiTM), 42300 Bandar Punck Alam, Selangor, Malaysia.
3Collaborative Drug Discovery Research (CDDR) Group,Pharmaceutical and Life Community of Research, Universiti Teknologi MARA, (UiTM),40450, Shah Alam, Selangor, Malaysia.
4Laboratory for Organic Reactivity, Discovery and Synthesis (LORDS), Research Center for the Natural and Applied Sciences, University of Santo Tomas, Espana Blvd., Manila 1015, Philippines.
Corresponding Author E-mail: tajudinali@salam.uitm.edu.my
DOI : http://dx.doi.org/10.13005/ojc/330128
Different derivatives based on the betulinic acid triterpenoidal scaffold were synthesized and characterized spectroscopically. The derivatives including synthetic intermediates were evaluated for their cytotoxic activity against the human colorectal carcinoma cell line (HCT116) using a colorimetric sulforhodamine assay (SRB). Among the synthesized compounds, compound 6 featuring a 20-keto seco-betulinic acid conjugated with a lactone benzyl ester through an amide linkage at C-28, exhibited the lowest IC50 value.
KEYWORDS:betulinic acid; seco-betulinic acid; lactone derivatives; ester derivatives; cytotoxicity; cancer
Download this article as:| Copy the following to cite this article: Ali M. T. M, Zahari H, Aliasak A, Lim S. M, Ramasamy K, Macabeo A. P. G. Synthesis, Characterization and Cytotoxic Activity of Betulinic Acid and Seco-Betulinic Acid Derivatives against Human Colorectal Carcinoma. Orient J Chem 2017;33(1). |
| Copy the following to cite this URL: Ali M. T. M, Zahari H, Aliasak A, Lim S. M, Ramasamy K, Macabeo A. P. G. Synthesis, Characterization and Cytotoxic Activity of Betulinic Acid and Seco-Betulinic Acid Derivatives against Human Colorectal Carcinoma. Orient J Chem 2017;33(1). Available from: http://www.orientjchem.org/?p=30371 |
Introduction
The pentacyclic lupane triterpenoid betulinic acid, 1 (3β, hydroxy-lup-20(29)-en-28-oic acid) is one of the first natural products identified from plants in 1788 [1]. It exhibits a number of biological activities including antitumor properties versus a panel of cancer cell lines. While previous studies suggest that betulinic acid is selectively cytotoxic against melanoma cell lines [2], it was also reported to exhibit activity against other types of human cancers including neuroblastoma, glioblastoma, medulloblastoma, Ewing tumor, leukemia as well as several carcinoma, i.e. head and neck, colon, breast, hepatocellular, lung, prostate, renal cell, ovarian or cervix carcinoma. Mechanistic studies indicate that it triggers the mitochondrial path to apoptosis [3], inhibits aminopeptidase N, an enzyme that is involved in the regulation of angiogenesis and overexpressed in several cancers [4-6], modulate activity of the transcription factor nuclear factor-κê (NF-êB), a key regulator of stress-induced transcriptional activation. Because it exhibits relative selective cytotoxicity against malignant over normal cells, betulinic acid is a promising anti-cancer agent.
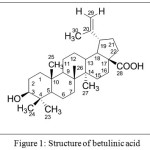 |
Figure 1: Structure of betulinic acid |
Several derivatization studies have been reported for betulinic acid leading to the synthesis of a library of betulinic acid congeners. Structurally, the triterpenoidal skeleton possesses three sites amenable for modification, including the C-3 hydroxyl, C-20 alkene, and C-28 carboxylic acid positions. While it holds a great potential to be developed as anti-tumor agent, it suffers from poor solubility in aqueous solutions and thus limiting its activity in vivo [7], [8].
In the light of the purported potential of betulinic acid and its derivatives as cancer chemopreventive agents, there exists a niche to develop new betulinic acid-inspired natural product analogs that may present better cytotoxic activity. In this paper we disclose the synthesis and evaluation of cytotoxicity versus human colorectal cancer cells of new betulinic acid and C-20 seco derivatives in vitro.
Experimental
Material
General. Infrared (IR) spectra (KBr) were recorded on a Thermo Nicolet Impact 6700 instrument (KBr pellet). 1H NMR spectra were recorded with a Bruker Ultra Shield 300 MHz spectrometer at 300 K, using TMS as an internal standard. Chemical shifts are expressed in parts per million (ppm, units). Coupling constants are in units of hertz (Hz). The spin multiplicities are indicated by the symbols s (singlet), d (doublet), dd (doublet of doublets), t (triplet), q (quartet), m (multiplet), and br (broad). MS spectra were recorded on a Agilent GC–7890A MS 5975 and Agilent LCMS QTof 6520. TLCs and preparative thin-layer chromatography were performed on silica gel GF/UV 254, and the chromatograms were performed on silica gel (100-200 mesh) visualized under UV light at 254 nm and 365 nm. All solvents were of reagent grade and, when necessary, were purified and dried by standard methods. Concentration of solutions after reactions and extractions involved the use of a rotary evaporator operating at a reduced pressure of ca. 20 Torr. Organic solutions were dried over anhydrous sodium sulfate.
Chemistry
In this work, a series of the new betulinic acid and C-20 seco derivatives were synthesized and characterized spectroscopically. Subsequently, the cytotoxicity of the derivatives including intermediates against human colorectal carcinoma cell line were explored, and the results are described below.
Reagents and conditions
(a) TFA, DCM, 0°C, 3 hrs, quant.
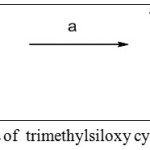 |
Scheme 1: Synthesis of trimethylsiloxy cyclohexene amine salt Click here to View scheme |
Reagents and Conditions
(a) DCM, HOBt (1.1 equiv.), HBTU, (1 equiv.) DiPEA (1.1 equiv, 0°C), 69%. (b) RuCl3.3H2O, NaIO4, CCl4:MeCN:H2O, 8 hrs, 0°C, 55%. (c) dry DMF, BnBr, NaHCO3, 24 hrs, rt, 72%
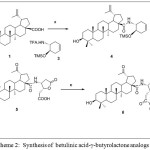 |
Scheme 2: Synthesis of betulinic acid-γ-butyrolactone analogs |
Reagents and Conditions
a) dry DMF, BnBr, NaHCO3, 24 hrs, rt. 68% (b) DCM, HOBt (1.1 equiv.), HBTU, (1 equiv.) DiPEA (1.1 equiv, 0°C), 85%
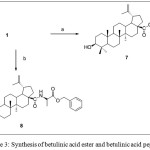 |
Scheme 3: Synthesis of betulinic acid ester and betulinic acid peptide |
Synthesis of Compounds 4 and 8
To a solution of betulinic acid 1 (30.0 mg, 0.07 mmol) in dry THF (2 mL) was added HOBt (11.2 mg, 1.1 equiv.), HBTU (30.0 mg, 1.0 equiv.) at 0ºC and trimethylsiloxycyclohexene amine TFA salt 3 or benzyl-D-alanine salt (1.1 equiv.) was added next. The ice bath was removed and the reaction mixture was stirred for 24 h at room temperature. The solvent was removed by rotary evaporator and the reaction mixture was redissolved in EtOAc (5 mL), and washed with 1M HCl (5 mL), 5% NaHCO3 (5 mL) and brine (5 mL). The organic layer was dried over by Na2SO4 and the solvent was evaporated under reduced pressure. The crude product was purified by column chromatography on silica gel (hexane: ethyl acetate, 15:1) to give products 4 as white solid or 8 (as white solid) in 69%or 85% yields, respectively.
(1R,3aS,5aR,5bR,9S,11aR)-9-hydroxy-5a,5b,8,8,11a-pentamethyl-1-(prop-1-en-2-yl)-N-((1R,6R)-6-trimethylsilyloxy)cyclohex-3-enyl)icosahydro-1H-cyclopenta[a]chrysene-3a-carboxamide (4)
Rƒ= 0.66 (SiO2, hexane/ethylacetate 4:1); Yields: 69%; m.p. 124- 126°C. 1H-NMR (,300 MHz CDCL3); δ= 5.23-5.35 (m, 2H, CH-Olefin), 4.73 (brs,1H, H-29), 4.61 (brs,1H, H-29), 3.66-3.75 (m, 1H, CHO), 3.53-3.65 ( m, 1H, CHN), 3.31-3.37 (m,1H, C-OH), 3.17-3.20 (m, 1H, CH, H-19)), 2.26-2.55 (m, 2H, CH2), 1.96-2.11(m, 2H, CH2), 1.69 (s,3H, CH3), 0.97 (s,3H, CH3), 0.96 (s,3H, CH3), 0.93 (s,3H, CH3) , 0.82 (s,3H, CH3), 0.75 (s,3H, CH3), 0.0 (s,9H, CH3-OTMS)
13C-NMR (75.5 MHz, CDCl3): δ = 172.0 (C=O), 150.12 (C=C, C-20), 124.1- 124.6 (C=C, Olefin), 107.42 (C=C, C-29), 83.71 (C-3), 78.8 (Cquart, cyclohexene), 78.9 (Cquart, cyclohexene), 57.34 (C-17), 55.48 (C-5), 51.16 (C-9), 50.6 (CH, Olefin), 50.41 (C-18), 46.1 (C-H, Olefin), 45.40 (C-19), 42.49 (C-14), 41.76 (C-8), 39.81 (C-1), 39.62 (C-4), 38.2 (C-13), 36.41 (C-10), 36.39 (C-22), 34.24 (C-7), 31.80 (C-16), 30.72 (C-15), 32.9 (CH2-Olefin), 29.9 (CH2-Olefin), 29.68 (C-21), 28.01 (C-23), 24.26 (C-12), 23.55 (C-11), 22.61 (C-
6), 19.7 (C-30), 19.31 (C-2), 16.71 (C-26), 16.45 (C-25), 16.23 (C-24), 15.05 (C-27), 0.00 (OTMS), MS [Cl, NH3]: m/z (%) 624.1 [M + H+], MS-ESI (Cl, NH3): Calculated for [C39H65NO3Si]: 623.6473, found 623.6460.
(2R)-benzyl 2-1R,3aS,5aR,5bR,9S,11aR)-9-hydroxy-5a,5b,8,8,11a-pentamethyl-1-(prop-1-en-2-yl)icosahydro-1H- cyclopenta[a]chrysene-3a-carboxamido) propanoate (8)
Rf = 0.3 (SiO2, hexane/ethylacetate 4:1); m.p. 117- 119oC.
1H-NMR (,300 MHz CDCL3); δ= 8.11-8.21 (m,1H,NH), 7.31-7.48 (m, 5H, Bn), 5.01-5.28 (d, 2H, J = 4 Hz, CH2Bn), 4.62 (brs, 1H, H-29), 4.75 (brs,1H, H-29), 4.11-4.48 (m, 1H, CHN), 2.96-3.01 (m, 1H, CH-OH), 3.15-3.31 (m,1H,CHCH3), 1.70 (m,3H, CH3), 1.42 (s,3H, CH3), 1.21 (s,3H, CH3), 1.10 (CH3), 0.97 (s,3H, CH3), 0.81 (s,3H, CH3), 0.69 (s,3H, CH3),
13C-NMR (75.5 MHz, CDCl3): δ = 175.8 (C=O), 172.0 (C=O), 150.9 (C=C, C-20), 136.4 (Cquart), 128.7- 128.5 (Ph), 109.5 (C=C, C-29), 78.9 (C-3), 65.7 (CH2Ph), 57.03 (C-17), 56.2 (CHN-ala), 55.42 (C-5), 51.05 (C-9), 50.48 (C-18), 45.40 (C-19), 42.40 (C-14), 41.60 (C-8), 39.81 (C-1), 39.88 (C-4), 38.5 (C-13), 35.41 (C-10), 36.30 (C-22), 34.24 (C-7), 31.80 (C-16), 30.62 (C-15), 29.38 (C-21), 28.2 (CH3-ala), 28.21 (C-23), 24.46 (C-12), 23.54 (C-11), 22.61 (C-6), 19.6 (C-30), 19.31 (C-2), 16.77 (C-26), 16.48 (C-25), 16.27 (C-24), 15.04 (C-27). IR (film) vmax cm-1 = 3410.4, 2924.01, 1690.3, 1166.3, 1591.1, 1241.7, MS-ESI (Cl, NH3): m/z. Calculated for [C40H59NO4] : 617.4444, found 618.3316 [M + H+].
Synthesis of compounds 5
To a solution of compound 4 (30 mg, 1 equiv.) in 4 ml of biphasic solution of CCl4:MeCN:H2O (1:1:2) in 50 ml round bottom flask was added 1.8 mg RuCl3.3H2O (8.3 % mol) at 0 ºC and the mixture was stirred vigorously for 5 minutes. NaIO4 (70 mg, 4.1 equiv.) was added portion wise and the reaction mixture was stirred for 8 h at 0-5 ºC. After completion of the reaction, the mixture was diluted with 10 ml of distilled water and extracted with 15 ml DCM (3x). The combined organic layer was dried over MgSO4 and the solvent removed using rotary evaporator. The crude product was purified by column chromatography on silica gel (ethyl acetate: methanol, 9:1) to furnish 5 as brown solid in 55% yield.
2-((2S,3R)-3-((1R,3aS,5aR,9S,11aR)-1-acetyl-9-hydroxy-5a,5b,8,8,11a-pentamethylicosahydro-1H-cyclopenta[a]chrysene-3a-carboxamido)-5-oxotetrahydrofuran-2-yl)acetic acid (5).
Rf = 0.60 (SiO2, hexane/ethylacetate 4:1); m.p. 151-153oC.
1H-NMR (300 MHz, CDCl3): 1H-NMR (,300 MHz CDCL3); δ= 4.93-5.03 (m,1H,CHN), 4.60-4.68 (m, 1H, CHCH2COOH), 3.30-3.36 (m,1H,CH-OH), 3.14-3.36 (m,1H,CH, H-19), 2.48-2.76 (m, 2H, CH2CN), 2.30- 2.49 (m, 2H, CH2-COO), 1.65 (s,3H,CH3), 1.40 (s,3H, CH3), 1.25 (s, 3H, CH3), 1.12 (s, 3H, CH3), 0.97 (s,3H, CH3), 0.81 (s,3H, CH3)
13C-NMR (75.5 MHz, CDCl3): δ = 171.9 (C=O), 169.6 (C=O), 149.71 (C=C, C-20)), 107.81 (C=C, C-29), 83.53 (C-3), 78.8 (C-OH, lactone), 66.8 (CH2, lactone), 56.95 (C-17), 55.35 (C-5), 50.51 (C-9), 49.65 (C-18), 45.88 (C-19), 42.36 (C-14), 40.78 (C-8), 38.7 (C-13), 38.88 (C-1), 38.7 (CH2-CO, lactone), 36.73 (C-10), 36.53 (C-22), 34.25 (CH2– lactone), 34.20 (C-7), 31.82 (C-16), 30.23 (C-15), 29.68 (C-21), 28.09 (C-23), 26.73 (C-12), 23.54 (C-11), 22.67 (C-6), 19.5 (C-30), 18.27 (C-2), 16.13 (C-26), 16.08 (C-25), 15.35 (C-24), 14.78 (C-27).
IR (film) vmax cm-1 = 2978.69-2933, 3420.82, 1714.86, 1255.07, 1499, 1367.79, 1738.28. MS [Cl, NH3]: m/z (%) 600.1 [M + H+], MS-ESI (Cl, NH3): m/z, Calculated for [C35H53NO7] : 599.3822, found 599.3460.38 [M+].
Synthesis of compounds 6
To a stirred solution of 5 (20 mg, 1 equiv.) in 4 ml dry DMF was added NaHCO3 (28 mg, 6 equiv.), followed by benzyl bromide (0.1 ml, 1.6 equiv.) sequentially at room temperature. The reaction mixture was stirred for 24 hours at room temperature. Then, the mixture was diluted with 4 ml EtOAc : H2O (1:1) solution and the solution was extracted. The aqueous layer extracted with EtOAc and the combine organic layer was dried over by anhydrous MgSO4 and concentrated reduced pressure to give yellow crude oil which was purified by column chromatography on silica gel (hexane: ethyl acetate, 6:4) to afford 6 as yellowish oil.
Benzyl 2-((2S,3R)-3-((1R,3aS,5aR,5bR,9S,11aR)-1-acetyl-9-hydroxy-5a,5b,8,8,11a-penamethylicosahydro-1H-cyclopenta[a]chrysene-3a-carboxamido}-5-oxotetrahydrofuran-2-yl)acetate (6).
Rf = 0.57 (SiO2, hexane/ethylacetate 4:1);
1H-NMR (,300 MHz CDCL3); δ= 8.11-8.21 (d, J= 4.2 Hz), 1H,NH), 7.27-7.33 (m, 5H, Bn), 5.15-5.18 (d, 2H, J= 4 Hz, CH2Bn), 4.90-4.95 (m, 1H, CHN), 4.69 (s,1H), 3.32-3.38 (m, 1H, CH-OH), 3.07-3.11 (m,1H,CH, H-19), 2.57-2.66 (m, 2H, CH2CN), 2.44-2.56 (m, 2H, CH2-COO) 1.75 (s,3H, CH3), 1.17 (s,3H, CH3), 1.11 (s, 3H, CH3), 0.98 (s,3H, CH3), 0.85 (s,3H, CH3), 0.66 (m,3H, CH3).
13C-NMR (75.5 MHz, CDCl3): δ = 177.2 (C=O), 173.3 (C=O), 149.78 (C=C, C-20), 132.4 (Cquart), 128.3- 128.5 (Ph), 107.21 (C=C, C-29), 81.22 (C-3), 78.5 (C-OH lactone), 68.2 (CH2Ph), 56.67 (C-17), 55.60 (C-5), 53.7 (CHN- lactone), 52.34 (C-9), 50.58 (C-18), 45.93 (C-19), 43.21 (C-14), 42.86 (C-8), 38.61 (C-1), 38.9 (C-13), 38.81 (C-4), 37.09 (C-10), 36.48 (C-H lactone), 36.41 (C-22), 34.44 (C-7), 31.91 (C-16), 29.68 (C-15), 29.11 (C-21), 28.95 (C-23), 25.70 (C-12), 23.64 (C-11), 22.66 (C-6), 19.65 (C-2),19.5 (C-30), 16.66 (C-26), 16.42 (C-25), 16.21 (C-24), 14.07 (C-27).
IR (film) vmax cm-1 = 2978.69-2933, 3420.82, 1714.86, 1255.07, 1499, 1367.79, 1738.28. MS-ESI (Cl, NH3): m/z Calculated for [C42H59NO7] : 689.4292, found 689.2452.
Synthesis of Compounds 7
To a stirred solution of 1 (100 mg, 1 equiv.) in 6 ml dry DMF was added NaHCO3 (110 mg, 6 equiv.), followed by benzyl bromide (60.1 mg, 1.6 equiv.) sequentially at room temperature. The reaction mixture was stirred for 24 hours at room temperature. Then, the mixture was diluted with 4 ml EtOAc–H2O (1:1) solution and the solution was extracted. The aqueous layer extracted with EtOAc and the combine organic layer was dried over by anhydrous MgSO4 and concentrated reduced pressure to give crude white solid product which was purified by column chromatography on silica gel (n-hex–EtOAc, 6:4) to afford 68% of a white solid 7.
(1R,3aS,5aR,5bR,9S,11aR)-benzyl 9-hydroxy-5a,5b,8,8,11a-pentamethyl-1-(prop-1-en-2-yl)icosahydro-1H-cyclopenta[a]chrysene-3a-carboxylate (7).
Rƒ = 0.5 ( SiO₂, hexane/ ethyl acetate), m.p. 162-164oC. ¹H NMR ( 300 MHz, CDCl3) : CDCl₃) : δ= 7.30-7.50 ( m, 5H, BnH), 5.12 (d, J= 2.2 Hz, CH2Ph), 4.73(brs,1H, H-29), 4.61(brs,1H, H-29), 3.26-3.32 (m, 1H, CHO), 3.03-3.15 ( m, 1H, CH, H-19), 2.26-2.55 (m, 2H, CH2), 1.69 (s,3H, CH3), 0.97 (s,3H, CH3), 0.96 (s,3H, CH3), 0.93 (s,3H, CH3) , 0.82 (s,3H, CH3), 0.75 (s,3H, CH3)¹³C NMR (75.5MHz, CDCl₃) : δ= 175.91 (C=O), 150.20 (C=C, C-20), 136.51 (Cquart), 128.7-128.2 ( Ph), 107.94 (C-29), 78.96 (C-3), 70.10 (CH2Ph), 57.04 (C-17), 55.40 (C-5), 50.59 (C-9), 49.99 (C-18), 46.58 (C-19), 42.50 (C-14), 40.82 (C-8), 38.88 (C-1), 38.7 (C-13), 38.60 (C-4), 37.22 (C-10), 36.32 (C-22), 34.92 (C-7), 31.53 (C-16), 29.69 (C-15), 28.36 (C-21), 27.99 (C-23), 25.71 (C-12), 23.66 (C-11), 21.94 (C-6), 19.66 (C-2), 19.6 (C-30), 16.15 (C-26), 16.11 (C-25), 15.35 (C-24), 14.84 (C-27). MS [Cl, NH3]: m/z (%) 547.3 [M + H+]. MS-ESI (Cl, NH3): m/z Calculated for [C37H54NO3] : 546.4073, found 547.2991 [M + H]+
Cytotoxicity Assay
HCT116 were seeded at 2,500 cells/ well (96 well plate) and exposed to respective compounds for 72 h before being subjected to fixation with 10% cold TCA and staining with 0.4% sulforhodamine. Plates were left to air-dry at room temperature and bound SRB was solubilised in 10 mM Tris base solution. The plate was read at 570nm using a spectrophotometer. A dose-response curve was plotted from which the IC50 value was determined. IC50 was defined as concentration of compound that corresponded to 50% cell viability when compared to control.
Results and Discussion
Scheme 1 shows the key starting materials for the synthesis of betulinic-γ-butyrolactone derivatives. NBoc trimethyl siloxycyclohexene 2 underwent deprotection Boc reaction by using trifluoro acetic acid (TFA, 30%) in DCM at 0°C to generate trimethyl siloxycyclohexene ammonium salt 3 in quantitative yield. The target compound, betulinic-γ-butyrolactone ester 6, was synthesized in three steps reaction starting from compound 3 [10]. The ammonium salt was incorporated with betulinic acid using standard peptide coupling procedure to afford betulinic-trimethylsiloxycyclohexene peptide 4 in 69% yield. Betulinic acid was preactivated by coupling reagent, HOBt/ HBTU and in the presence of strong base DIPEA.
Subsequently, peptide 4 underwent lactonization by ruthenium tetraoxide, RuO4 in the presence of sodium periodate as an oxidant to give the corresponding betulinic-γ-butyrolactone carboxylic acid 5. The RuO4 catalytic oxidative system was generated in situ from 8.3 % of RuCl3.3H2O and 4.1 equivalent of NaIO4 in biphasic solution of CCl4:MeCN:H2O with ratio of (1:1:2). The conversion of cyclohexene silyl ether with co-solvent system was completed in 8 hours at 0°C.
In accordance to previous study on lactonisation reaction involving oxidative cleavage reaction of trimethyl siloxycyclohexene scaffold, the configuration of both asymmetric centre on the butyrolactone skeleton is cis configuration [9]. The possibility of cis confirmation may be due to the presence of ruthenium tetraoxide which could create interactions on the similar face between nitrogen lone pair to metal and chelation of metal and carboxylate oxygen atom.
Benzylic protection of carboxylic acid 5 was envisioned by treating 5 with benzyl bromide in the present of sodium hydrogen carbonate as base to give betulinic-γ-butyrolactone ester 6. In addition, betulinic acid underwent benzylation reaction after being treated with benzyl bromide, BnBr to afford 7. Betulinic-D-alanine peptide could be synthesized by applying peptide synthesis procedure furnished peptide 8 in a good yield [10].
Cytotoxic Activity
The inhibitory activity of the synthesized compounds against HCT116 was evaluated by cytotoxic assay. In general, compound 1 had emerged as the potent anticancer agent, with an IC50 of 3.0 μg/mL. This was followed by compound 7, 8, and 4, with IC50 value 25.33, 36.67 and 50.00 μg/mL respectively. The IC50 of compound 5 and 7 were beyond the range of concentration tested. Betulinic-g-butyrolactone carboxylic acid, 5 yielded an IC50 > 100 μg/mL . Its benzyl ester derivative 6, however, yielded IC50 at least four times lower (IC50 value = 25.3 μg/mL). Eventhough none of the compounds exhibited comparable activity to that of positive control (5-FU), the results showed that incorporation of betulinic acid with γ-butyrolactone ester moiety contributed to the inhibition of HCT116.
Conclusion
In summary, several novel of betulinic-γ-butyrolactone and betulinic acid derivatives were synthesized and tested for their in vitro cytotoxic activities against a human colorectal carcinoma cell lines (HCT116). The results of preliminary biological activity showed that two synthesized compounds (6 and 8) possessed potential cytotoxicities. Further structure exploration especially on the aromatic butyrolactone ester side chains are in progress in our lab.
Acknowledgement
The authors are thankful to the Universiti Teknologi MARA Malaysia (grant no: 600-RMI/DANA 5/3/RIF 690/2012 and FRGS grant no: 600-RMI/FRGS 5/3(3/2013) for financial support. Mohd Salleh Rofie from Integrative Pharmacogenomic Institute (i-PROMISE), Universiti Teknologi MARA for mass analysis.
References
- Alakurtti S, Makela T, Koskimies S, Yli-Kauhaluoma J. Eur. J. Pharm. Sci., 2006,.29, 1–13.
CrossRef - Pisha E, Chai H, Lee IS, Chagwedera TE, Farnsworth NR, Cordell GA, Beecher CW, Fong HH, Kinghorn AD, Brown DM, et al. Nat. Med., 1995.,1, 1046–1051 .
CrossRef - Galluzzi L, Larochette N, Zamzami N, Kroemer G. Oncogene. 2006.,25, 4812–4830 .
CrossRef - Kwon HJ, Shim JS, Kim JH, Cho HY, Yum YN, Kim SH, Yu J. Jpn. J. Cancer Res., 2002., 93, 417–425 .
CrossRef - Melzig MF, Bormann H. Planta Med., 1998.,64, 655–657 .
CrossRef - Sjostrom H, Noren O, Olsen J. Adv. Exp. Med. Biol., 2000.,477, 25–34 .
CrossRef - Gomathi P, Girma T, Mebrahtom G, Biruk S, Michael G, Aman K, Gereziher G. Archives of Applied Science Research, 2014., 6, 47-58.
- Ali M T M, Jasmani D, Y Yasin. 2005. Institute of Research, Development and Commercialization, Universiti Teknologi MARA 2005.
- Ali M T M, Macabeo A P G, Wan B, Franzblau, S, Reiser O. In Abstracts of Papers of The American Chemical Society, 2013.,245 .
- Masdar N D, Mahmud A H, Ali M T M, Ismail S N A S, Tajuddin R, Saim N, Md Jani A M. Advanced Materials Research, 2015.,1.,105, 231-236 .

This work is licensed under a Creative Commons Attribution 4.0 International License.
![]()
A New Edition of Web of Science
Journal Impact Factor
2022: 0.5
Five Year: 0.8
Journal is Indexed in

Cabells Whitelist
![]()


