Ftir, Ft-Raman and Dft Calculations of 5-Nitro-1,3-Benzodioxole
C.Yohannan Panicker1*, Hema Tresa Varghese2 and Y. Sheena Mary2
1Department of Physics, TKM College of Arts and Science, Kollam, Kerala (India). 2Department of Physics, Fatima Mata National College, Kollam, Kerala (India).
FTIR and FT-Raman spectrum of 5,-benzo-1,3-benzodioxole were recorded and analyzed. The vibrational wavenumbers were examined theoretically using the Gaussian03 set of quantum chemistry codes. The experimental frequencies are in agreement with the calculated (B3LYP) scaled values. The predicted infrared intensities and Raman activities are reported.
KEYWORDS:FTIR; FT-Raman; DFT; benzodioxole
Download this article as:| Copy the following to cite this article: Panicker C. Y, Varghese H. T, Mary Y. S. Ftir, Ft-Raman and Dft Calculations of 5-Nitro-1,3-Benzodioxole. Orient J Chem 2012;28(2). |
| Copy the following to cite this URL: Panicker C. Y, Varghese H. T, Mary Y. S. Ftir, Ft-Raman and Dft Calculations of 5-Nitro-1,3-Benzodioxole. Available from: http://www.orientjchem.org/?p=23536 |
Introduction
The benzodioxole ((methylenedioxy)benzene) group is present in a number of endothelin receptor antagonists. Endothelins are 21-amino acid bicyclic peptides originally isolated from the supernatant of cultured porcine endothelial cells1. There have been many studies about the molecular conformations of indan-like benzene fused ring molecules2-4. These molecules have received considerable attention due to their interesting conformational properties. Laane et al. reported the infrared, Raman, electronic UV absorption, and laser induced fluorescence excitation spectra of two oxygen containing indan-like molecule, phthalan5-7 and 1,3-benzodioxole8,9. 1,3-benzodioxole has been studied extensively by several spectrographic and theoretical methods owing to its propensity for large amplitude motion10-12. It is believed that the molecule has a nonplanar structure in its electronic ground state. While the aromatic ring is planar, the five member ring is puckered, with the oxygen atoms and the CH2 group on opposite side of the aromatic plane. In the present work, the vibrational spectroscopic analysis of 5-nitro-1,3-benzodioxole is reported, experimentally and theoretically.
Experimental
The FT-IR spectrum was recorded on a DR/Jasco FT-IR 6300 spectrometer in KBr pellets, number of scans 16, resolution 2 cm-1. The FT-Raman spectrum was obtained on a BRUKER RFS 100/S, Germany. For excitation of the spectrum the emission of a Nd:YAG laser was used, excitation wavelength 1064 nm, maximal power 150 mW. One thousand scans were accumulated with a total registration time of about 30 min. The spectral resolution after apodization was 2 cm-1.
Computational Details
Calculations of the title compound were carried out with Gaussian03 software program13 using the B3LYP/6-31G* basis sets to predict the molecular structure and vibrational wavenumbers. The DFT hybrid B3LYP functional tends also to overestimate the fundamental modes; therefore scaling factors have to be used for obtaining a considerably better agreement with experimental data. Therefore, a scaling factor of 0.9613 was uniformly applied to the DFT calculated wavenumbers14. The assignment of the calculated wavenumbers is aided by the animation option of MOLEKEL program, which gives a visual presentation of the vibrational modes15,16.
Results And Discussion
The observed IR, Raman and calculated (scaled) wavenumbers and the assignments are given in Table 1. In nitro compounds, υasNO2 are located in the region 1580 ± 80 cm-1. The nitro benzene derivatives17 shows υasNO2 in the region 1535 ± 30cm-1 and the 3-nitropyridine at 1530 ± 20cm-1. In the present case, υasNO2 are obtained at 1596 theoretically and at 1609 cm-1 in IR spectrum. In nitro compounds, υsNO2 are located in the region 1400 ± 40 cm-1. For the title compound, υsNO2 is observed at 1427 cm-1 (DFT), 1437 cm-1 (IR) and 1430 cm-1 (Raman). In the aromatic nitro compounds, bands are usually seen at 855 ± 40cm-1 (NO2 scissors deformation δNO2), 760 ± 30cm-1 (NO2 out of plane wag ωNO2), 580 ± 30cm-1 (NO2 in-plane rock ρNO2) and 70 ± 20cm-1 (τNO2). In the present case, the observed values for δNO2 are 917 (DFT), 920 (IR) cm-1; ωNO2 is 803 cm-1 (DFT), ; ρNO2 are 666 (DFT) and τNO2 is at 21cm-1 (DFT). Panicker et al. [18] reported the NO2 deformation bands at 800, 724, 534 cm-1 (theoretically) and 809, 727, 717, 524 cm-1 (experimentally). Sundaraganesan et al.19 reported the NO2 deformation bands at 839, 744 and 398cm-1 experimentally and 812, 716,703 and 327 cm-1 theoretically.
For methylene groups20, the CH2 vibrations are observed in the region 2800-3000, 1200-1400, 875-1150 and 600-950 cm-1. The vibrations of the CH2 group (the asymmetric stretch υasCH2, symmetric stretch υsCH2, the scissoring vibration and wagging vibration) appear in the regions 2940-3005, 2870-2940, 1420-1480 and 1320-1380 cm-1, respectively17, 21. The stretching bands of the CH2 groups are observed at 2925 cm-1 in the IR spectrum and at 2992, 2942 cm-1 theoretically for the title compound. According to literature22 scissoring mode of the CH2 group give rise to characteristic band near 1485 cm-1 in IR and Raman spectra. This mode is observed at 1489 cm-1 in the IR spectrum and at 1474 cm-1 theoretically. The wagging mode is observed at 1378 cm-1 in the Raman spectrum and at 1372 cm-1 theoretically. The twisting and rocking vibrations of the CH2 group appear in the region17 of 1200-1280 and 740-900 cm-1, respectively. These modes are also assigned. For the title compound the twisting vibration is observed at 1115 cm-1 in the IR spectrum and at 1100 cm-1 theoretically. The rocking deformation is assigned at 907 cm-1 theoretically.
The asymmetric and symmetric C-O-C stretching vibrations are expected to appear at 1150-1250 and 1000-1050 cm-1 [22]. In the present case the DFT calculations give these modes at 1224, 1160, 1046 and 1027 cm-1. The bands at 1171, 1066, 1036 (IR) and 1051, 1028 cm-1 (Raman) are assigned as C-O-C symmetric stretching modes. The C-O-C stretching modes are reported at 1250, 1073 cm-1 for 2-mercaptobenzoxazole23, 1263, 1055 cm-1 for 5-methyl-2-(p-fluorophenyl)benzoxazole24.
The existence of one or more aromatic rings in a structure is normally readily determined from the C–H and C=C–C related vibrations. The C–H stretching occurs above 3000 cm-1 and is typically exhibited as a multiplicity of weak to moderate bands, compared with the aliphatic C–H stretch25. In the present case, the DFT calculations give the υCH modes at 3145, 3134 and 3110 cm-1. The band observed at 3119 cm-1 in the IR spectrum is assigned as υCH mode of the benzene ring. The benzene ring possesses six ring stretching vibrations, of which the four with the highest wavenumbers (occurring near 1600, 1580, 1490 and 1440 cm-1) are good group vibrations17. In the absence of ring conjugation, the band near 1580 cm-1 is usually weaker than that at 1600 cm-1. The fifth ring stretching vibration which is active near 1335 ± 35 cm-1 a region which overlaps strongly with that of the CH in-plane deformation and the intensity is in general, low or medium high17. The sixth ring stretching vibration or ring breathing mode appears as a weak band near 1000 cm-1 in mono, 1,3-di and 1,3,5-trisubstituted benzenes. In the other wise substituted benzene, however, this vibration is substituent sensitive and difficult to distinguish from the ring in-plane deformation. For tri-substituted phenyl ring the υPh modes17 are seen in the region 1640 – 1250 cm-1 and these modes are observed at 1632, 1551, 1516, 1381, 1345 cm-1 in the IR spectrum, 1620, 1333 cm-1 in the Raman spectrum and at 1619, 1563, 1515, 1395, 1336 cm-1 (DFT) theoretically. In asymmetric tri-substituted benzenes, when all the three substituents are light, the wavenumber interval of the breathing mode26 is between 500 and 600 cm-1. When all the three substituents are heavy, the wavenumber appears above 1100 cm-1. In the case of mixed substituents, the wavenumber is expected26 to appear between 600 and 750 cm-1. For the title compound the phenyl ring breathing mode is assigned at 796 cm-1 theoretically. Mary et al.27 reported the ring breathing mode of tri-substituted benzene ring at 733 cm-1 in the IR spectrum and at 738 cm-1 theoretically. The in-plane bending δCH modes17 of the phenyl ring are expected above 1000 cm-1. For the tri-substituted benzene ring these modes are observed at 1120, 1036 cm-1 in IR, 1128, 1028 cm-1 in Raman and at 1144, 1103, 1027 cm-1 theoretically. The CH out-of-plane deformations17 are observed between 1000 and 700 cm-1. Generally the CH out-of-plane deformations with the highest wavenumbers have a weaker intensity than those absorbing at lower wavenumbers. These γCH modes are observed at 872, 810, 719 (IR), 809, 725 (Raman), 864, 806, 721 cm-1 (DFT). The in-plane and out-of-plane substituent modes of the phenyl ring are also identified and assigned (Table 1).
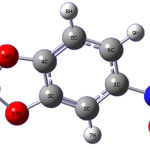 |
scheme 1 Click here to View scheme |
Conclusion
The FTIR and FT-Raman spectra of 5-nitro-1,3-benzodioxole were recorded and analyzed. The harmonic vibrational wavaenumbers were calculated theoretically using Gaussian03 set of quantum chemistry codes. The calculated wavenumbers (B3LYP) agree well with the observed wavenumbers. The small differences between experimental and calculated vibrational modes are observed. It must be due to the fact that hydrogen bond vibrations present in the crystal lead to strong perturbation of the infrared wavenumbers and intensities of many other modes. Also we state that the experimental results belong to solid phase and theoretical calculations belong to gaseous phase.
Table 1: Calculated (scaled) wavenumbers, IR, Raman bands and assignments
| B3LYP/6-31G*υ(cm-1) | IR intensity | Raman activity | IRυ(cm-1) | Ramanυ(cm-1) | Assignments |
| 3145 | 6.64 | 54.46 | υCH | ||
| 3134 | 2.48 | 77.13 | υCH | ||
| 3110 | 1.89 | 92.31 | 3119 | υCH | |
| 2992 | 40.89 | 163.43 | υasCH2 | ||
| 2942 | 115.42 | 250.34 | 2925 | υsCH2 | |
| 1619 | 25.45 | 11.34 | 1632 | 1620 | υPh |
| 1596 | 15.65 | 73.75 | 1609 | υasNO2 | |
| 1563 | 189.45 | 6.23 | 1551 | υPh | |
| 1515 | 1.32 | 38.51 | 1516 | υPh | |
| 1474 | 302.11 | 0.49 | 1489 | δCH2 | |
| 1427 | 20.57 | 33.63 | 1437 | 1430 | υsNO2 |
| 1395 | 3.13 | 12.10 | 1381 | υPh | |
| 1372 | 13.89 | 12.77 | 1378 | ωCH2 | |
| 1336 | 428.93 | 404.83 | 1345 | 1333 | υPh |
| 1260 | 375.73 | 26.55 | 1270 | 1272 | υCN |
| 1224 | 40.89 | 6.75 | υCOC | ||
| 1160 | 0.01 | 9.54 | 1171 | υCOC | |
| 1144 | 4.87 | 2.59 | δCH | ||
| 1103 | 10.66 | 0.97 | 1120 | 1128 | δCH |
| 1100 | 16.73 | 1.37 | 1115 | τCH2 | |
| 1046 | 22.51 | 17.81 | 1066 | 1051 | υCOC |
| 1027 | 145.43 | 3.68 | 1036 | 1028 | υCOC,δCH |
| 936 | 32.58 | 8.05 | 930 | 932 | γCH |
| 917 | 0.56 | 2.32 | 920 | δNO2 | |
| 907 | 29.56 | 7.10 | ρCH2 | ||
| 864 | 24.85 | 1.08 | 872 | γCH | |
| 806 | 13.30 | 2.52 | 810 | 809 | γCH |
| 803 | 1.55 | 6.40 | 805 | ωNO2 | |
| 796 | 33.34 | 23.60 | υPh | ||
| 721 | 31.63 | 2.05 | 719 | 725 | γCH |
| 715 | 3.60 | 11.80 | γPh | ||
| 678 | 0.79 | 0.23 | 686 | δPh(X) | |
| 666 | 10.98 | 2.14 | ρNO2 | ||
| 570 | 5.99 | 7.14 | 574 | γPh(X) | |
| 544 | 0.13 | 0.10 | 546 | δPh(X) | |
| 528 | 3.82 | 0.92 | γPh(X) | ||
| 411 | 0.58 | 0.50 | δPh(X) | ||
| 394 | 2.81 | 3.46 | δCX(X) | ||
| 339 | 0.01 | 2.62 | δCX(X) | ||
| 316 | 0.36 | 2.33 | γCX(X) | ||
| 224 | 0.13 | 2.51 | γCX(X) | ||
| 203 | 1.18 | 1.15 | 208 | δCX(X) | |
| 119 | 4.59 | 0.02 | γCX(X) | ||
| 63 | 0.17 | 0.17 | 75 | tPh | |
| 21 | 6.43 | 0.69 | tNO2 |
υ-stretching; δ-in-plane deformation; γ-out-of-plane deformation; ω-wagging; ρ-rocking; t-torsion; as-asymmetric; s-symmetric; Ph-phenyl ring; X-substituent sensitive.
Refernces
- Yanagisawa, M., Kurihara, H., Kimura, S., Tomobe, Y., Kobayashi, M., Mitsui, Y., Goto, K., .Masaki, T.A., Nature 332: 411 (1988).
- Laane, J., J. Phys. Chem. 104A: 7715 (2000).
- Caminati, W., Damiani, D., Favero, L.B., Mol. Phys. 79: 699 (1993).
- Leal, L.A., .Alonso, J.L., Lister, D.G., Lopez, J.C., Mol. Phys. 81: 1205 (1994).
- Klots, T., Sakurai, S., Laane, J., J. Chem. Phys. 108: 3531 (1998).
- Sakurai, S., Meinander, N., Laane, J., J. Chem. Phys. 108: 3537 (1998).
- Bondoc, E., Sakurai, S., Morris, K., Chiang, W.Y., Laane, J., J. Chem. Phys. 112: 6700 (2000).
- Sakurai, S., Meinander, N., Morris, K., Laane, J., J. Am. Chem. Soc. 121: 50 (1999).
- Laane, J., Bondoc, E., Sakurai, S., Morris, K., Meinander, N., Choo, J., J. Am. Chem. Soc. 122: 2628 (2000).
- Duckett, J.A., Smithson, T.L., Wieser, H., Chem. Phys. Lett. 64: 261 (1979.
- Caminati, W., Melandri, S., Corebelli, G., Favero, L.B., Meyer, R., Mol. Phys. 80: 1297 (1993).
- Sakurai, S., Meinander, N., Morris, K., Laane, J., J. Am. Chem. Soc. 121: 5056 (1999).
- Frisch, et al., M.J., Gaussian 03, Revision C.02 Gaussian, Inc., Wallingford CT (2004).
- Foresman, J.B., in: Frisch, E., (Ed.), Exploring Chemistry with Electronic Structure Methods: A Guide to Using Gaussian”, Pittsburg, PA (1996).
- Flukiger, P., Luthi, H.P., Portmann, S., Weber, J., MOLEKEL 4.3, Swiss Centre for Scientific Computing, Manno, Switzerland (2000-2002).
- Portmann, S., Luthi, H.P., Chimia 54: 766 (2000).
- Roeges, N.P.G., A Guide to the Complete Interpretation of the Infrared spectra of organic structures, Wiley, NewYork (1994).
- Panicker, C.Y., Varghese, H.Y., Ushakumari, L., Ertan, T., Yildiz, I., Granadeiro, C.M., Nogueira, H.I.S., Mary. Y.S., J. Raman Spectrosc. 41: 381 (2010).
- Sundaraganesan, N., Ayyappan, S., Umamaheshawari, H., Joshua, B.D., Spectrochim. Acta 66: 17 (2007).
- Socrates, G., IR characteristic group frequencies, John Wiley and Sons, New York (1981).
- Colthup, N.B., Daly, L.H., Wiberly, S.E., Introduction to Infrared and Raman Spectroscopy, second ed., Academic Press, New York (1985).
- Silverstein, R.M., Bassler, G.C., Morril, T.C., Spectrometric Identification of Organic Compounds, Ed. 5, John Wiley and Sons Inc., Singapore (1991).
- Bigotto, A., Pergolese, B., J. Raman Spectrosc. 32: 953 (2001).
- Anto, P.L., Panicker, C.Y., Varghese, H.T., Philip, D., Temiz-Arpaci, O., Gulbas, B.T., Yildiz, I., Spectrochim Acta 67: 744 (2007).
- Coates, J., Meyers, R.A., Introduction to Infrared Spectrum, A Practical Approach, Chichester, John Wiley and Sons Ltd, (2000).
- Varsanyi, G., Assignments for Vibrational Spectra of Seven Hundred Benzene Derivatives, New York, Wiley (1974).
- Mary, Y.S., Varghese, H.T., Panicker, C.Y., Ertan, T., Yildiz, I., Arpaci, O.T., Spectrochim. Acta 71A: 566 (2008).

This work is licensed under a Creative Commons Attribution 4.0 International License.
![]()
A New Edition of Web of Science
Journal Impact Factor
2022: 0.5
Five Year: 0.8
Journal is Indexed in

Cabells Whitelist
![]()


