Dft Study on 4(5)-Imidazole-Carbaldehyde-N(5)-Phenylthiosemicarbazone (Imtph): Nmr Shielding Tensors, Thermodynamic Parameters, Nbo Analysis, Molecular Electrostatic Potential (Mep), Homo and Lumo Studies
Masoome Sheikhi, Mohammad Mahmoodi Hashemi*, Majid Monajjemi
Department of Chemistry, College of Basic Sciences, Tehran Science and Research Branch, Islamic Azad University, Tehran, Iran.
DOI : http://dx.doi.org/10.13005/ojc/300146
Article Received on :
Article Accepted on :
Article Published : 04 Mar 2014
The density functional theory (DFT) calculations at the level of B3LYP/6-31G was carried out on the structure 4(5)-Imidazole-carbaldehyde-N(5)-phenylthiosemicarbazone (ImTPh) in gas phase using Gaussian 03. Dipole moment (Debye), energy of structure formation (HF; kcal/mol) and point group, NMR parameters such as isotropic shielding (σiso) and anisotropic shielding (σaniso), σ11, σ22 and σ33 obtained. Also thermodynamic properties and natural bond orbitals (NBO) were calculated. Besides, the frontier molecular orbital (FMO) analysis and the molecular electrostatic potential (MEP) of the compound were investigated by theoretical calculations.
KEYWORDS:DFT; ImTPh; NMR parameter; Thermodynamic parameter; NBO; FMO; MEP.
Download this article as:| Copy the following to cite this article: Sheikhi M,Hashemi M .M, Monajjemi M. Dft Study on 4(5)-Imidazole-Carbaldehyde-N(5)-Phenylthiosemicarbazone (Imtph): Nmr Shielding Tensors, Thermodynamic Parameters, Nbo Analysis, Molecular Electrostatic Potential (Mep), Homo and Lumo Studies. Orient J Chem 2014;30(1) |
| Copy the following to cite this URL: Sheikhi M,Hashemi M .M, Monajjemi M. Dft Study on 4(5)-Imidazole-Carbaldehyde-N(5)-Phenylthiosemicarbazone (Imtph): Nmr Shielding Tensors, Thermodynamic Parameters, Nbo Analysis, Molecular Electrostatic Potential (Mep), Homo and Lumo Studies. Orient J Chem 2014;30(1). Available from: http://www.orientjchem.org/?p=2279 |
Introduction
Imidazole is an organic compound with the formula C3H4N2. This aromatic heterocyclic is classified as an alkaloid. Imidazole refers to the parent compound whereas imidazoles are a class of heterocycles with similar ring structure but varying substituents. The imidazole nucleus is well known to play an important role in living organisms since it is incorporated into the histidine molecule and many other important biological systems. Imidazole derivatives show various pharmacological activities such as antifungal and anti-bacterial activity1, anti inflammatory activity and analgesic activity2, antitubercular activity3, anti depressant activity, anti cancer activity4, antiviral activity5 and antileishmanial activity. Due to their antifungal properties imidazole-derived compounds have been used in agriculture as effective ingredients for controlling plants pests. Imidazole derivatives are employed in the control of spoilage microorganisms or organisms potentially harmful to man, in the protection of wood against fungi and also in food storage6. Thiosemicarbazones7,8 and hydrazones9 are reported as compounds which present significant antifungal activity. The imidazole-derived thiosemicarbazones were prepared by reacting of 4(5)-imidazole-carboxaldehyde, 4-(1H-imidazole-1-yl)-benzaldehyde or 4-(1H-imidazole-1-yl)- acetophenone with the suitable thiosemicarbazide using methanol as solvent10. In the present work we carried out theoretical calculations on one of the imidazole-derived thiosemicarbazones, that is named 4(5)-Imidazole-carbaldehyde-N(5)-phenylthiosemicarbazone (ImTPh).
Theoretical calculations
We have carried out quantum theoretical calculations of ImTPh using DFT (B3LYP) method with 6-31G basis set by the Gaussian 03 program11. We calculated 1H and 13C NMR chemical shifts of ImTPh using B3LYP/6-31G level within GIAO approach. Moreover was studied thermodynamic parameters of ImTPh using methods listed, and obtained the energy (ΔE), enthalpies (∆H), Gibbs free energy (∆G), entropies (S) and constant volume molar heat capacity (Cv) of derivatives12. Some electronic properties such as energy of the highest occupied molecular orbital (EHOMO), energy of the lowest unoccupied molecular orbital (ELUMO), energy gap (Eg; Δ) between LUMO and HOMO, atomic charges, dipole moment (μ) and Point group were determined. The optimized molecular structures, HOMO and LUMO surfaces were visualized using GaussView 03 program13. We also studied electronic structures of ImTPh using Natural Bond Orbital (NBO) analysis using level of B3LYP/6-31G. The main listing of NBOs, displaying the form and occupancy of the complete set of NBOs that span the input AO space and for each orbital gives the type of orbital and the occupancy14.
Discussion
The optimized molecular structure of ImTPh was calculated using Gaussian 03 software. The optimized geometrical parameters, such as Dipole moment (Debye), energy of structure formation (HF; kcal/mol) and point group, obtained using B3LYP method and 6-31G as the basis set that are listed in Table 1. As shown in Table 1, ImTPh has C1 point group symmetry and Dipole moment (μ) value is 5.4185. Also the energy of structure formation (HF) of ImTPh is negative.
| Table 1. Dipole moment, HF and Point group of ImTPh obtained using B3LYP/6-31G level. | ||
|
HF (kcal/mol) |
Dipole moment(Debye) | Pointgroup |
| -688676.535 | 5.4185 | C1 |
NMR Parameters
In this section we report and analyze NMR shielding tensors of 1H, 13C, 15N-NMR such as isotropic shielding (σiso) and anisotropic shielding (σaniso), σ11, σ22, σ33 of ImTPh, which obtain using level of B3LYP/6-31G in gas phase. The chemical shielding anisotropy (CSA) tensors provide important information about electronic environment of the nuclei, which depends on the molecular geometry. The results of our studies are listed Table 2. According to Table 2, isotropic shielding value (σiso) and anisotropic shielding value (σaniso) for C1, C2, C4, C6, C9 atoms are a positive values. Of carbon atoms mentioned, C9 has the lowest isotropic shielding and the highest anisotropic shielding value. The isotropic shielding value of N3 and N7 (-C=N) is negative and anisotropic shielding for these atoms is the highest value. The isotropic and anisotropic shielding values of hydrogen atoms are positive. The H20 has the lowest isotropic shielding and the highest anisotropic shielding value. Therefore H20 is the most acidic hydrogen.
| Table 2. NMR shielding tensors values of ImTPh obtained using B3LYP/6-31G level. | |||||
|
isoσ |
anisoσ |
11σ |
22σ |
33σ |
|
|
C1 |
68.109 | 105.736 | 7.598 | 58.129 | 138.600 |
|
C2 |
63.816 | 114.798 | -4.365 | 55.464 | 140.347 |
|
C4 |
61.335 | 97.839 | -8.789 | 66.234 | 126.561 |
|
C6 |
68.474 | 100.343 | -12.944 | 82.998 | 135.369 |
|
C9 |
15.892 | 121.787 | -128.044 | 78.637 | 97.084 |
| N3 | -40.283 | 409.594 | -268.882 | -84.746 | 232.779 |
| N5 | 102.382 | 121.185 | 8.110 | 115.864 | 183.172 |
| N7 | -71.636 | 380.997 | -353.853 | -43.418 | 182.361 |
| N8 | 79.316 | 93.185 | 6.673 | 89.836 | 141.440 |
| N11 | 123.777 | 105.585 | 40.554 | 136.610 | 194.167 |
|
H19 |
25.353 | 3.797 | 22.976 | 25.199 | 27.884 |
|
H20 |
23.747 | 9.631 | 17.025 | 24.049 | 30.168 |
|
H21 |
25.080 | 5.337 | 22.776 | 23.826 | 28.638 |
|
H22 |
25.696 | 3.834 | 22.484 | 26.352 | 28.252 |
|
H23 |
26.829 | 8.131 | 19.613 | 28.624 | 32.250 |
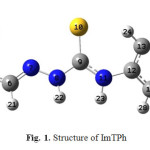 |
Fig. 1. Structure of ImTPh |
Frequency calculations
Thermodynamic parameters such as the relative energy (ΔE), standard enthalpies (ΔH), entropies (ΔS), Gibbs free energy (ΔG) and constant volume molar heat capacity (Cv) values of ImTPh were obtained using B3LYP/6-31G level. The values are listed in Table 3 showed that relative energy, Gibbs free energy and standard enthalpies of ImTPh are negative that we found this structure is stable.
|
Table 3. The calculated thermodynamic parameters of ImTPh using B3LYP/6-31G level
|
||||
|
E(Kcal/mol)∆ |
ΔG(Kcal/mol) | ΔH(Kcal/mol) |
S(cal/molK) |
CV(cal/molK) |
| -688530.795 | -688568.278 | -688530.202 | 127.707 | 55.106 |
NBO analysis
In accordance with the simple bond orbital picture, each bonding NBO is introduced as an orbital formed from two directed valence hybrids (NHOs) hA, hB on atoms A and B, with corresponding polarization coefficients cA, cb. Table 3 show share of orbitals contribute in the bonds (BD for 2-center bond). As shown in Table 4, in the N5-H20 bond, BD= 0.8603sp2.19 + 0.5097s reported. Thus polarization coefficients of the N5-H20 bond N5= 0.8603 and H20= 0.5097 reported, that sizes of these coefficients show hybrid of N5 is more important in the formation of the N5-H20 bond. Also in N8-H22 and N11-H23 bonds, polarization coefficient values of N8 and N11 is larger than H22 and H23. The of polarization coefficient values of H20, H22 and H23 is 0.5097, 0.5366 and 0.5344, respectively. This show that share of contribute H20 in bond N5-H20 is lower than share of H22 and H23 in N8-H22 and N11-H23 bonds, respectively.
| Table 4. Calculated natural bond orbitals (NBO) and the polarization coefficient for each hybrid in N-H bonds of ImTPh.
|
||||||
|
BD* |
|
BD |
||||
|
B
|
A |
A-B |
B |
A |
A-B |
|
|
s(-0.8603) |
sp2.19 (0.5097) |
N5-H20 |
s(0.5097) |
sp2.19 (0.8603) |
N5-H20 |
|
|
s(-0.8438) |
sp2.59 (0.5366) |
N8-H22 |
s(0.5366) |
sp2.37 (0.8438) |
N8-H22 |
|
|
s(-0.8452) |
sp2.59 (0.5344) |
N11-H23
|
s(0.5344) |
sp2.90 (0.8452) |
N11-H23
|
|
| * Antibonding | ||||||
Frontier Molecular Orbital (FMO) Analysis
The EHOMO, ELUMO and HOMO-LUMO energy gap (Eg; Δ) of ImTPh was calculated using the B3LYP method and 6-31G basis set. The properties of molecular orbitals such as energy and frontier electron density are important and are used to determine the reactive position. According to Fig. 2, the HOMO energy and the LUMO energy (ELUMO) of ImTPh is -5.61 eV and -1.74 eV, respectively. Also Fig.2 and spectrum DOS (Fig. 3) show that energy gap of ImTPh is 3.87 eV. As shown in Fig. 2, the HOMO is focused mainly around and sulfur atom and imidazole ring.
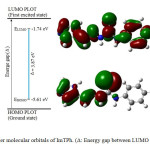 |
Fig. 2. Frontier molecular orbitals of ImTPh. (Δ: Energy gap between LUMO and HOMO). Click here to View Figure |
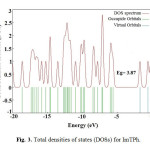 |
Fig. 3. Total densities of states (DOSs) for ImTPh. Click here to View Figure |
Molecular Electrostatic Potential
Molecular electrostatic potential (MEP) is relevant to the electronic density and it to specify the locations sites of electrophilic and nucleophilic15. MEP methodology studies the electronic distribution in the molecule16. The optimized geometry of ImTPh to indicate sites of electrophilic and nucleophilic, MEP was calculated using B3LYP method and 6-31G basis set. As shown in Fig. 4, nitrogen atom of imidazole ring (N3) has maximum electronic density (red color). Therefore N3 is main negative center. Also lowest electron density is observed for hydrogen atoms bound to nitrogen (blue color).
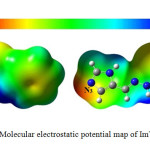 |
Fig. 4. Molecular electrostatic potential map of ImTPh. Click here to View Figure |
The atomic charge of atoms of ImTPh is shown in Fig. 5. As can be seen in Fig. 5, H20 (Hydrogen attached to the nitrogen in the imidazole ring) has the most positive charge.
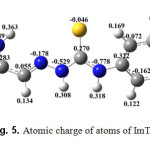 |
Fig. 5. Atomic charge of atoms of ImTPh Click here to View Figure |
Conclusion
In the present work, we have performed theoretical analyses of 4(5)-Imidazole-carbaldehyde-N(5)-phenylthiosemicarbazone (ImTPh).The following conclusions are obtained from the current study: The point group symmetry, Dipole moment (μ) and energy of structure formation (HF) of ImTPh is C1, 5.4185 and -688676.535, respectively. As regards hydrogen bonded to nitrogen of imidazole ring (H20) has the lowest valu of σiso and σaniso, therefore H20 the most acidic hydrogen of ImTPh. According to the thermodynamic parameters, we find that amount of Gibbs free energy (ΔG), standard enthalpies (ΔH) and internal thermal energy (ΔE) of ImTPh is negative value, therefore ImTPh is stable structure. Among hydrogens bonded to nitrogen, H20 in bond N5-H20 has the lowest polarization coefficient. This suggests that H20 in imidazole ring is the most acidic hydrogen of ImTPh. According to FMO analysis, the HOMO is focused mainly around and sulfur atom and imidazole ring. As shown MEPs, the maximum electron density is located on the nitrogen atom (N3) imidazole ring. Finally, Our results confirm the biological properties of compounds containing the imidazole ring among 4(5)-Imidazole-carbaldehyde-N(5)-phenylthiosemicarbazone (ImTPh).
References
- Shingalapur R. V., Hosamani K. M., Keri R. S., European Journal of Medicinal Chemistry, 44, 4244-4248 (2009).
- Puratchikodya A., Doble M., Bioorganic & Medicinal Chemistry. 15, 1083-1090 (2007).
- Pandey J., Tivari V. K., Verma S. S., Chaturvedi V., Bhatnagar S., Sinha S., Gaikwad A. N., Tripathi R. P., European Journal of Medicinal Chemistry, 44, 3350-3355 (2009).
- Ramla M. M., Mohamed A., Abdel-Momen M., Kharmry E., Hoda I., Diwani E., Bioorg. Med. Chem., 14, 7324-7332 (2006).
- Tonelli M., Simone M., Tasso B., Novelli F., Boido V., Bioorganic & Medicinal Chemistry, 18, 2937-2953 (2010).
- Melander C., Cavanagh J., Ritchie D. F., Rogers S. A., Robert W., Inhibition and dispersion of biofilms in plants with imidazole-triazole derivatives, WIPO Patent 2010077603, 8 July (2010).
- Pervez H., Iqbal M. S., Tahir M. Y., Nasim F., Choudhary M. I., Khan K. M., J. Enzym. Inhib. Med. Chem., 23, 848–854 (2008).
- Beraldo H., Gambino D., Min. Rev. Med. Chem., 4, 159–165 (2004).
- Rollas S., Küçükgüzel Ş. G., Molecules, 12, 1910–1939 (2007).
- Débora C. R., Angel A. Recio., Jeferson G. D., Nayane F. S., Camila F. V., Isolda C.M., Jacqueline A. T., Heloisa B., Molecules, 18, 12645-12662 (2013)
- M.J. Frisch et al., Gaussian 03, revision B03, Gaussian Inc., Pittsburgh, PA, 2003.
- Monajjemi M., Afsharnezhad S., Jaafari M. R., Abdolahi T., Nikosade A., Monajemi H., Russ. J. Phys. Chem. A, 81, 1956–1963 (2007).
- Frisch A., Nielson A. B., Holder A. J., GAUSSVIEW User Manual, Gaussian Inc., Pittsburgh, PA, 2000.
- Seyed Katouli S. A., Sheikhi M., Seikh D., Tayyari S. F., Orient. J. Chem., 29, 1121-1128 (2013).
- Luque F. J., Lopez J. M., Orozco M., Theor. Chem. Acc., 103, 343–345 (2000).
- Ramachandran S., Velraj G., Rom. Journ. Phys., 58, 305-318 (2013).

This work is licensed under a Creative Commons Attribution 4.0 International License.
![]()
A New Edition of Web of Science
Journal Impact Factor
2022: 0.5
Five Year: 0.8
Journal is Indexed in

Cabells Whitelist
![]()


