Design and Synthesis of N-(2-{[(2-Hydroxy-naphtalen-1-yl)- phenyl-methyl]-amino}-ethyl)-3,4-dinitro-benzamide
Lauro Figueroa-Valverde1 *, Francisco Diaz-Cedillo2 , Elodia Garcia-Cervera1 ,Eduardo Pool-Gomez1 , Maria Lopez-Ramos1 and Abelardo Camacho-Luis2
1Lab. Farmacoquímica, Facultad de Ciencias Químico-Biológicas, Universidad Autónoma de Campeche, Av. Agustín Melgar, Col Buenavista C.P.24039 Campeche Cam., México. 2Escuela Nacional de Ciencias Biológicas del Instituto Politécnico Nacional. Prol. Carpio y Plan de Ayala s/n Col. Santo Tomas, México, D.F. C.P. 11340. 3 Facultad de Medicina y Nutrición, Centro Investigación en alimentos y nutrición. Universidad Juárez del Estado de Durango. Av. Universidad s/n esq. Fanny Anitua, Zona centro. Durango, México.
In this study, a new naphtol derivative was synthesized. The first stage, involved preparation of 1-[(2-Amino-ethylamino)-phenylmethyl]-naphthalen-2-ol (4) using a three-component system (β-naphtol [1], benzaldehyde [2], and ethylenediamine [3]), followed by the reaction of 4 with 3,5- dinitrobenzoic acid (5) to form N-(2-{[(2-Hydroxy-naphtalen-1-yl)-phenyl-methyl]-amino}-ethyl)- 3,5-dinitro-benzamide (6) using a carbodiimide derivative or boric acid. The second stage, involves the synthesis of N-(2-aminoethyl)-3,5-dinitrobenzamide (7) by reaction of 5 with 3 using a carbodiimide derivative. Finally, the compound 6 was also synthetized using the three-component system (compound 7, benzaldehyde and â-naphtol). The structure of all compounds obtained was confirmed by elemental analysis, spectroscopy and spectrometry data. In conclusion, this method offers some advantages such as good yields, simple procedure, low cost, and ease of workup.
KEYWORDS:Naphtol derivative; Boric acid; Carbodiimide
Download this article as:| Copy the following to cite this article: Figueroa-Valverde L, Diaz-Cedillo F, Garcia-Cervera E, Pool-Gomez E, Lopez-Ramos M, Camacho-Luis A. Design and Synthesis of N-(2-{[(2-Hydroxy-naphtalen-1-yl)- phenyl-methyl]-amino}-ethyl)-3,4-dinitro-benzamide. Orient J Chem 2013;29(1). |
| Copy the following to cite this URL: Figueroa-Valverde L, Diaz-Cedillo F, Garcia-Cervera E, Pool-Gomez E, Lopez-Ramos M, Camacho-Luis A. Design and Synthesis of N-(2-{[(2-Hydroxy-naphtalen-1-yl)- phenyl-methyl]-amino}-ethyl)-3,4-dinitro-benzamide. Available from: http://www.orientjchem.org/?p=11932 |
Introduction
There are several methods reported for synthesis of aromatic-condensed derivatives; for example, the synthesis of naphtyl ketone by the reaction of o-alkynylbenzaldehydes with alkynes using AuCl3 as catalyst1. Other reports indicate the tandem pummerer Diels-Alder sequence for the preparation of α-thiosubstituted naphthalene derivatives2. Additionally, other studies3 showed the synthesis of 1,8-diphenylnaphtalene and 1-Iodo-8-phenylnaphtalene by the reaction of lithium diphenylcuprate and aryl halides. Other studies reported by Ganapathy and Viswanathan4 shown the synthesis of polysubstituted naphthalene derivatives through gallium trichloride catalyzed by alkyne-aldehyde coupling. In addition, some carbamato-alkyl-naphtol derivatives5,6 have been synthetized by condensation of β-naphtol, aromatic aldehyde and methyl carbamate in ionic liquid media. Other studies indicate that the compound N-(2,4-Dibromonaphthyl)benzamide was prepared by benzoylation of 2,4-Dibromonaphthylamine in pyridine7. Additionally, other naphtalenbenzamide derivative (N-(3-mercapto-5-(naphthalen-1-yl)-4H-1,2,4-triazol-4-yl)benzamide) was synthetized by the reaction of potassium 2-(2-naphthoyl)hydrazinecarbodithioate with N-amino-arylcarboxamides in ethanol to reflux8. Recently, was synthetized a naphthalene-benzamide derivative (N-(3-(1-Methoxy-naphthalen-2-yl)-2,2-dimethylpropyl)-2-benzamide) by the reaction of benzoic acid with 3-(1-1 Methoxy-naphtalen-2-yl)-2,2-dimethyl-propylamine in methanol at room temperature9. All these experimental results show several procedures that are available for synthesis of naphthalene derivatives; nevertheless, expensive reagents and special conditions are required. Analyzing these data, in this study a naphthalene derivative (N-(2-{[(2-Hydroxy-naphtalen-1-yl)-phenyl-methyl]-amino}-ethyl)-3,4-dinitrobenzamide) was synthetized using several chemical strategies.
Experimental
General methods
The compound 1-[(2-Amino-ethylamino)-phenyl-methyl]-naphthalen-2-ol (1) was synthetized by amethod previously reported10. The other compounds were purchased from Sigma-Aldrich Co.,Ltd. The melting points for the different compounds were determined on anElectrothermal (900 model). Infrared spectra (IR) were recorded using KBrpellets on a Perkin Elmer Lambda 40 spectrometer. 1H and 13C NMRspectra were recorded on a Varian VXR-300/5 FT NMR spectrometer at300 and 75.4 MHz in CDCl3 using TMS as internal standard. EIMS spectrawere obtained with a Finnigan Trace GCPolaris Q. spectrometer.Elementary analysis data were acquired from a Perkin Elmer Ser. IICHNS/0 2400 elemental analyzer.
Synthesis of 1-[(2-Amino-ethylamino)-phenyl-methyl]-naphthalen-2-ol.(4)
A solution of β-naphtol (100 mg, 0.69 mmol), benzaldehyde (105 µl, 1.03mmol), ethylenediamine (92 µl, 1.38 mmol) in 10 ml of ethanol wasstirring for 72 h to room temperature. The reaction mixture was evaporatedto a smaller volume. After the mixture was diluted with water and extractedwith chloroform. The organic phase was evaporated to dryness underreduced pressure, the residue was purified by crystallization frommethanol:water (4:1) yielding 75 % of product, m.p. 52-54 oC; IR ṽ =3530, 3330, 3310 cm-1; 1H NMR (300 MHz, CDCl3) dH: 2.88 (t, 2H, J =11 6.0 Hz), 3.08 (t, 2H, J = 6.0 Hz), 3.72 (broad, 3H), 5.75(s, 1H), 6.88 (m,12 2H), 7.05 (m, 1H), 7.11 (m, 2H), 7.20 (m, 1H), 7.41-7.54 (m, 3H), 7.68-7.75 (m, 2H) ppm. 13C NMR (75.4 Hz, CDCl3) dC:42.20 (C-15), 52.98(C-14), 55.53 (C-12), 114.39 (C-9), 121.17 (C-8), 123.57 (C-6), 125.65(C-2), 126.21 (C-7), 127.39 (C-20), 128.01 (C-4), 128.22 (C-10), 130.02(C-18, C-22), 130.06 (C-5), 130.88 (C-19, C-21), 137.07 (C-17), 138.35(C-3), 152.11 (C-1) ppm. EI-MS m/z: 292.07 (M+, 11). Anal. Calcdfor:C19H20N2O : C, 78.05; H, 6.89; N, 9.58; O, 5.47. Found: C, 78.02; H, 6.85.
Synthesis of N-(2-{[(2-Hydroxy-naphtalen-1-yl)-phenyl-methyl]-amino}-ethyl)-3,5-dinitro-benzamide. (6)
Method A
Compound 4 (100 mg, 0.34 mmol) was added to a solution of3,5-dinitrobenzoic acid (110 mg, 0.52 mmol) and 42 mg boric acid (0.68mmol) in 15 ml of acetonitrile:water (3:1) and the mixture was stirred atroom temperature for 72 h. The solvent was then removed under vacuumand the crude product was purified by crystallization from methanol:hexane:water (3:2:1) yielding 45% of product (6). m.p.: 252-254 oC; IR:ṽ = 3512, 3312, 1,638, 1,380 cm-1; 1H NMR (300 MHz, CDCl3) dH: 2.78(t, 2H, J = 6.0 Hz), 3.08 (t, 2H, J = 6.0 Hz), 5.10 (s, 1H), 5.48 (broad,3H), 6.86 (m, 2H), 7.06 (m, 1H), 7.13 (m, 2H), 7.20 (m, 1H), 7.40-7.57(m, 3H), 7.72 (m, 2H), 8.92-9.02 (d, 3H) ppm. 13C NMR (75.4 Hz, CDCl3)dC:42.30 (C-15), 53.54 (C-14), 55.52 (C-12), 114.40 (C-9), 120.02 (C-28),121.18 (C-8), 123.55 (C-6), 126.61 (C-7), 126.76 (C-2), 127.39 (C-22),127.50 (C-26, C-30), 128.11 (C-4), 128.14 (C-10), 129.25 (C-24, C-20),129.28 (C-5), 131.02 (C-21, C-23), 137.57 (C-25), 138.18 (C-19), 138.35(C-3), 146.97 (C-27, C-29), 152.10 (C-1), 160.87 (C-17) ppm. EI-MS m/z:486.53(M+, 17). Anal. Calcdfor C26H22N4O6: C, 64.19; H, 4.56; N, 11.52;O, 19.73. Found: C, 64.02; H, 4.51.
Method B
Compound 4 (100 mg, 0.34 mmol) was added to a solution of 3,5-dinitrobenzoic acid (110 mg, 0.52 mmol) and 1-ethyl-3-(3-dimethylaminopropyl)-carbodiimide hydrochloride (130 mg, 0.68 mmol) in 10 ml of methanol. After,the mixture was stirring at room temperature for 72 h. The solvent wasthen removed under vacuum and the crude product was purified bycrystallization from methanol-hexane-water to give naphtol derivate (6)(68% yield) similar 1H NMR and 13C NMR data were obtained comparedwith method A product.
Synthesis of N-(2-aminoethyl)-3,5-dinitrobenzamide. (7)
A solution of 5 (100 mg, 0.47 mmol), ethylenediamine (63 μL, 0.94 mmol)and 1-ethyl-3-(3-dimethylaminopropyl)-carbodiimide hydrochloride (180mg, 0.94 mmol) in 20 ml of acetonitrile/methanol/water (3:2:1) wasstirring for 72 h to room temperature. The reaction mixture was evaporatedto a smaller volume. After, the mixture was diluted with water and extractedwith chloroform. The organic phase was evaporated to dryness underreduced pressure, the residue was purified by crystallization frommethanol:water (4:1) yielding 75 % of product, m.p. 198 oC; IR: ṽ = 3332,1630, 1384 cm-1; 1H NMR (300 MHz, CDCl3) dH: 3.02 (t, 2H, J = 6.0 Hz),3.46 (t, 2H, J = 6.0 Hz), 4.46 (broad, 3H), 8.92-9.04 (d, 3H) ppm. 13C NMR(75.4 Hz, CDCl3) dC: 42.60 (C-15), 43.58 (1 C-16), 120.68 (C-6), 125.70 (C-2, C-4), 136.50 (C-3), 147.04 (C-1, C-5), 158.76 (C-9) ppm. EI-MS m/z:254.12(M+, 17). Anal. Calcd. for C9H10N4O5: C, 42.52; H, 3.97; N, 22.04;O, 31.47. Found: C, 42.50; H, 3.90.
Synthesis of N-(2-{[(2-Hydroxy-naphtalen-1-yl)-phenyl-methyl]-amino}-ethyl)-3,5-dinitro-benzamide. (6)
A solution of β-naphtol (100 mg, 0.69 mmol), benzaldehyde (105 µl, 1.03mmol), compound 7 (180 mg, 0.70 mmol) and 1-ethyl-3-(3-dimethylaminopropyl)-carbodiimide hydrochloride in 15 mL of acetonitrile:water(3:1) was stirring for 72 h to room temperature. The reaction mixture wasevaporated to a smaller volume. After the mixture was diluted with waterand extracted with chloroform. The organic phase was evaporated todryness under reduced pressure, the residue was purified by crystallizationfrom methanol:water (4:1) yielding 75 % of product. The signals IR, 1HNMR and 16C NMR of naphtol derivative (6) were confirmed byspectroscopic analyses as in the first method mentioned above.
Results and Discussion
It is important to mention that many procedures for formation of naphtolderivatives are available in the literature. Nevertheless, despite their widescope, these procedures suffer from several drawbacks; some reagents areof limited stability, and preparation can be dangerous11,12. Therefore, inthis study we report a straightforward route for synthesis of new naphtolderivative (6). The first step involves preparation of 4 using the threecomponent system (β-naphtol, benzaldehyde and ethylenediamine) inethanol (Scheme 1). The infrared spectra of 4 show bands at 3530 forhydroxyl group; at 3330 for primary amino group and 3310 for secondaryamino group. The 1H NMR spectrum of 4 shows signals at 2.88 and 3.08ppm for protons involved in the arm present in the compound 4; at 5.75ppm for hydrogen of methylene which is bound to both phenyl and naphtolgroups. Other signal at 3.72 ppm for both amino and hydroxyl groups werefound. Finally, the spectrum contains several signals at 6.88, 7.11 and 7.20ppm for phenyl group; at 7.05, 7.41-7.75 ppm for naphtol group. The 13CNMR spectrum contains peaks at chemical shifts of 42.20 and 52.98 ppmfor the carbons of the methylenes involved in the arm of 4. A signal at55.53 ppm for methylene bound to phenyl group and naphtol group wasfound. In addition, several signals at 114.39-126.21, 128.01-128.22, 130.06and 138.35-152.11 ppm for carbons involved in the naphtol group; at127.39, 130.02, 130.88-137.07 ppm for carbons of phenyl group werefound. In addition, the presence of 4 was further confirmed from massspectrum which showed a molecular ion at m/z 292. 07.
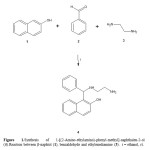 |
Figure 1: |
The second step involves the synthesis of 6 by reaction of 4 with 3,5-dinitrobenzoic acid (5) resulting in amide bond formation. It is important tomention that many procedures for the formation of amide groups areknown in the literature. The most widely practiced method employscarboxylic acid chlorides as the electrophiles which react with the aminogroup in the presence of an acid scavenger13. Despite its wide scope, theformer protocol suffers from several drawbacks; most notable are thelimited stability of many acid chlorides and the need for hazardous reagentsfor their preparation (thionyl chloride)14. Therefore, in this study twodifferent methods for amide formation were employed: in the first one thetechnique reported by Pingwah15 for boric acid catalyzed amidation ofcarboxylic acids and amines (method B) was used; in the second one weused a carbodiimide derivate (method A) as catalyst16 for the amidebond formation to obtain 6. In this study, the use of carbodiimide derivativewas found to result in higher yields compared to the amide bond formingwith method B. The results of infrared spectroscopic analyses showedcharacteristic bands at 3512 for hydroxyl group; at 3312 for secondaryamino group; at 1638 for amide group and 1380 for nitro group. The1H NMR spectrum of 6 shows signals at 2.78 and 3.08 ppm for protonsinvolved in the spacer arm between naphtol group and dinitrophenyl group;at 5.10 ppm for hydrogen of methylene which is bound to both phenyl andnaphtol groups. In addition, several signals at 6.86, 7.13 and 7.20 ppm forphenyl group; at 7.06 and 7.40-7.72 ppm for naphtol group; at 8.92-9.02ppm for dinitrophenyl group were found. Finally, the spectrum contains asignal characteristic at 5.48 ppm for amide, amino and hydroxyl groups. Itis important to mention that the 1H NMR spectra of the secondary amidesare usually more complex than the primary amides due to the presence of asubstituent bonded to the amide nitrogen atom. These substituents producea much wider range of chemical shifts for the amide proton which may, inaddition, display coupling to aliphatic groups bonded to it. The chemicalshifts of aliphatic groups bonded to the carbonyl group are similar to thoseobserved for the primary amides, while those groups bonded to the nitrogenresonate at slightly lower field than the corresponding amines17.
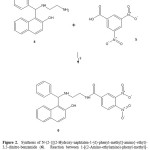 |
Figure 2: Click here to View figure |
On the other hand, the 13C NMR spectrum of 6 contains peaks at chemicalshifts of 42.30 and 53.54 ppm for the carbons of the methylenes of spacerarm between naphtol group and dinitrophenyl group; at 55.52 ppm forcarbon bound to both phenyl group and naphtol group. In addition, othersignals at 114.40, 121.18-126.76, 128.11-128.14, 129.28, 138.35 and152.10 ppm for carbons involved in naphtol group; at 120.02, 127.39,129.25, 131.02 and 138.18 ppm for phenyl group; at 125.50, 137.57, and146.97 for dinitrophenyl group ppm; at 160.87 ppm for amide group werefound. Finally, the presence of 6 was further confirmed from massspectrum which showed a molecular ion at m/z 486.53.
On the other hand, in the search of another alternative for synthesis of 6;the compound 7 was synthetized which later was used as reagent to form 6.Therefore, the preparation of 7 was made by the reaction of 5 withethylenediamine (3) using a carbodiimide derivative as catalyst. Theinfrared spectra of 7 show bands at 3332 for primary amino group; at 1680for amide group and at 1384 for nitro group. The 1H NMR spectrum of 7shows signals at 3.02 and 3.46 ppm for protons involved in the arm boundto phenyl group. Other signals at 4.46 ppm for both amide and aminogroups; at 8.92-9.04 ppm for phenyl groups were found. The 13C NMRspectrum of 7 contains peaks at chemical shifts of 42.60 and 43.58 ppm forthe carbons of the methylenes of arm bound to phenyl group; at 12.68-147.04 ppm for phenyl group; at 158.76 ppm for amide group. Finally, thepresence of 7 was further confirmed from mass spectrum which showed amolecular ion at m/z 254.12.
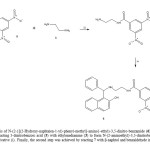 |
Figure 3: |
On the other hand, the second stage for synthesis of 6 was developed usingthe three-component system (β-naphtol, benzaldehyde and compound 7) inethanol (Scheme 4, see). It is important to mention that with this methodthe yielding was higher in comparison with the first stage. The signals IR,1H NMR and 13C NMR of naphtol derivative (6) were confirmed byspectroscopic analyses as in the first method mentioned above.In conclusion, in thisstudy are reported several strategies for thesynthesis of the naphthol derivative (6).It isimportant to mention that the methods used arehighly versatile and the yield is good.
References
- Asao N., Takahashi K., Lee S.,Kasahara T. and Yamamoto Y.,J. Am.Chem. Soc.,124:12650-12651 (2002).
- Cochran J. andPadwa A.,Tetrahedron Lett.,30:3495-3498 (1995).
- House H.,Koepsell D. and Campbell W.,J. Org. Chem.,37:1003-1011 (1972).
- Ganapathy S. andViswanathan A.,Angew. Chem.,114:2242-2245 (2002).
- Dabiri M.,Delbari A. ansBazgir A.,Heterocycles.,7:543-547 (2007).
- In-Soo B.,Yoo-Shin K. and Yong-Tae P.,Bull. Korean Chem. Soc.,24:916-920 (2003).
- Desai N.,Shihora P.,Rajpara K., Joshi V.,Vaghani H.,Satodiya H. andDodiya A.,Med.Chem. Res.,2011: DOI 10.1007/s00044-011-9833-8
- PradidpholN.,Kongkathip N.,Sittikul P.,Boonyalai N. andKongkathip, B.,Eur. J. Med. Chem.,49:253-270 (2012).
- Noji M., Nakajima M. and Koga K.,Tetrahedron Lett.,35:7983-7984 (1994).
- Figueroa-Valverde L., Díaz-Cedillo F., García-Cervera E. and Pool-Gómez E. Design and Synthesis of Three Naphtol Derivativesusing the Three Component System. Oriental J. Chem., 28(3):1085-1090 (2012).
- Kobayashi S.,Moriwaki M. andHachiya I.,Tetrahedron Lett.,37:2053-2056 (1996).
- Reza H.,Hosseinian A. andGhashang M.,Tetrahedron Lett.,49:5804-5806 (2008).
- Medvedeva A., Andreev M.,Safronova L. andSarapulova G.,Arkivoc.,ix:143-149 (2001).
- Levin D.,Org. Process.Res. Dev.,1:182-184 (1997).
- Pingwah T.,Org.Synt. 81:262-267 (2005).
- DeSilva N.,Am. J.Respir.Cell. Mol. Biol.,29:757-770 (2003).
- Figueroa-Valverde L., Díaz-Cedillo F., Tolosa L., Maldonado G. andCeballos-Reyes G., J.Mex.Chem. Soc.,50:42-45 (2006).

This work is licensed under a Creative Commons Attribution 4.0 International License.
![]()
A New Edition of Web of Science
Journal Impact Factor
2022: 0.5
Five Year: 0.8
Journal is Indexed in

Cabells Whitelist
![]()


