Quantum chemical study on the corrosion inhibition property of some heterocyclic azole derivatives
N. Anusuya1, P. Sounthari2, J. Saranya2, K. Parameswari2, S. Chitra2*
1Department of Chemistry,RVS Faculty of Engineering, Coimbatore, India
2Department of Chemistry, PSGR Krishnammal College for Women, Coimbatore, India.
Corresponding Author E-mail: rajshree1995@rediffmail.com
DOI : http://dx.doi.org/10.13005/ojc/310355
Article Received on :
Article Accepted on :
Article Published : 20 Aug 2015
Quantum chemical calculations based on density functional theory (DFT) method were performed on heterocyclic azole derivatives as corrosion inhibitors for mild steel in acid media to investigate the relationship between molecular structure of the inhibitors and the corresponding inhibition efficiencies (%). Quantum chemical parameters most relevant to their potential action as corrosion inhibitors have been calculated in the non-protonated and protonated forms in aqueous phase for comparison. Results obtained in this study indicate thatin acidic media, both the protonated and non-protonated forms of the azoles represent the better actual experimental situation.
KEYWORDS:azole derivatives; mild steel; density functional theory; corrosion inhibitors; protonation; adsorption
Download this article as:| Copy the following to cite this article: Anusuya N, Sounthari P, Saranya J, Parameswari K, Chitra S. Quantum chemical study on the corrosion inhibition property of some heterocyclic azole derivatives. Orient J Chem 2015;31(3). |
| Copy the following to cite this URL: Anusuya N, Sounthari P, Saranya J, Parameswari K, Chitra S. Quantum chemical study on the corrosion inhibition property of some heterocyclic azole derivatives. Orient J Chem 2015;31(3). Available from: http://www.orientjchem.org/?p=10321 |
Introduction
The dissolution rate of steel during cleaning, pickling, scaling and etching is quite high in acidic medium, the inhibition of such dissolution may be achieved with organic compounds containing π-electrons and /or hetero atoms (i.e., N, O and S) which can be adsorbed on the metal surface(1, 2). In order to evaluate the synthesized compounds as corrosion inhibitors and to design novel inhibitors, more research work has been concentrated on the studies of the relationship between structural characteristics of organic compounds and inhibiting effects. It has been suggested that the most effective factors for the inhibiting effects are the electronegative atoms, unsaturated bonds and the plane conjugated systems including all kinds of aromatic cycles, of which they can offer special active electrons or vacant orbital to donate or accept electrons(3, 4).
Experimental means are useful in explaining the corrosion inhibition mechanism but they are often expensive and time consuming since it is always based on large scale trial and error experiments. However, ongoing computer hardware and software advances have opened the door for powerful use of theoretical chemistry in corrosion inhibition research (5). Quantum chemical calculations can complement the experimental investigations or even predict with confidence some experimentally unknown properties. Recently, there has been increasing use of the density functional theory (DFT) methods as a theoretical tool in elucidating the mechanism of corrosion inhibition of organic compounds by several researchers (6-8). The advancement in methodology and implementations has reached a point where predicted properties of reasonable accuracy can be obtained from DFT calculations (9). However, despite enormous literature available on the use of DFT in understanding the corrosion inhibition mechanism, information on the use of statistical analysis as a tool in correlating the experimentally determined inhibition efficiencies and the calculated quantum chemical parameters in the non-protonated and protonated forms are scarce.
This paper reports the correlation between the observed inhibition efficiency oftwo azole derivatives used as corrosion inhibitors with their calculated quantum chemical parameters both in the non-protonated and in the protonated forms using statistical tool. The calculations of global reactivity indices of the inhibitors such as the localization of frontier molecular orbitals, EHOMO, ELUMO, energy gap (ΔE), dipole moment (µ), hardness (η), softness (S), electrophilicity index (ω) andthe fractions of electrons transferred (ΔN) using DFT at B3LYP/6-31G (d) basis set level were used to explain the electron transfer mechanism between the inhibitor molecules and the mild steel surface.
Experimental
Synthesis of Inhibitors
Synthesis of Chalcones (Ia)
An ethanolic solution of acetophenone (0.01mol) and 2-pyridine carboxaldehyde in the presence of catalytic amount of 40% KOH was stirred for 3hours at room temperature. It was then poured over crushed ice and the product formed was crystallized from ethanol.
Synthesis of Azole Derivatives (IIa&b)
A mixture of chalcone (0.02mol), hydrazine hydrate/hydroxylaminehydrochloride (0.02mol) and glacial acetic acid (10ml) in ethanol (25ml) were refluxed overnight. The mixture was concentrated by distilling out the solvent under reduced pressure and poured into ice. The precipitate obtained was filtered, washed and recrystallized from ethanol (10).The reactions are presented in scheme 1. The chemical structures and their abbreviations for the heterocyclic azole derivatives chosen for the study are presented in Table 1.
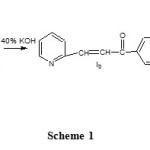 |
Scheme 1 Click here to View scheme |
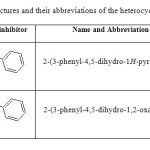 |
Table 1: Chemical structures and their abbreviations of the heterocyclic azole derivatives Click here to View table |
Evaluation of Inhibition of the Azole Derivatives
Weight loss Method
The gravimetric method (weight loss) is probably the most widely used method for inhibition assessment. The simplicity and reliability of the measurement offered by the weight loss method is such that the technique forms the baseline method of measurement in corrosion monitoring program.
Table 2: Experimental inhibition efficiency obtained from gravimetric measurements for the corrosion of mild steel in 1MH2SO4at 303 K
|
Name of the inhibitor |
Inhibitor concen. (mM) |
Weight loss (g) |
Inhibition efficiency (%) |
Corrosion rate (gcm-2 hr-1) |
Surface coverage (θ) |
|
Blank |
0.2059 |
– |
13.34 |
– |
|
|
PPP |
0.05 0.1 0.25 0.35 0.5 1.0 |
0.0513 0.0472 0.0382 0.0279 0.0212 0.0165 |
75.08 77.08 81.45 86.45 89.70 91.99 |
3.32 3.06 2.47 1.81 1.37 1.07 |
0.7508 0.7708 0.8145 0.8645 0.8970 0.9199 |
|
POP |
0.05 0.1 0.25 0.35 0.5 1.0 |
0.0605 0.0529 0.0392 0.0289 0.0218 0.0172 |
70.62 74.31 80.96 85.96 89.41 91.65 |
3.92 3.43 2.54 1.87 1.41 1.11 |
0.7062 0.7431 0.8096 0.8596 0.8941 0.9165 |
The initial weight of the polished specimen was taken. The solutions were taken in 100ml beakers and the specimens were suspended in triplicate into the solution using glass hooks. Care was taken to ensure the complete immersion of the specimen. After a period of three hours, the mild steel samples were taken out, washed with distilled water, dried and weighed to the accuracy of four decimals. From the initial and final mass of the specimen, (i.e before and after immersion in the solution) the loss in weight was calculated. The experiment was repeated for various concentrations of the synthesized inhibitors.
Table 3: Calculated quantum chemical parameters for the inhibitors in the neutral form obtained using DFT at the B3LYP/6-31G (d) basis set in aqueous phase.
|
Quantum chemical parameters |
PPP |
POP |
| Total energy (eV) |
-705.57 |
-725.41 |
| Dipole moment (debye) |
5.0689 |
3.1648 |
| EHOMO (eV) |
-5.5106 |
-6.2202 |
| ELUMO (eV) |
-1.3072 |
-1.1028 |
| ΔE gap (eV) |
4.1944 |
5.1174 |
| Ionization potential (eV) |
5.5106 |
6.2202 |
| Electron affinity (eV) |
1.3072 |
1.1028 |
| Electronegativity (eV) |
3.4044 |
3.6615 |
| Global hardness (eV) |
2.0972 |
2.5587 |
| Global softness |
0.4768 |
0.3908 |
| ΔN |
0.8572 |
0.6524 |
The inhibition efficiency, corrosion rate and surface coverage were calculated from the weight loss results using the formulas,
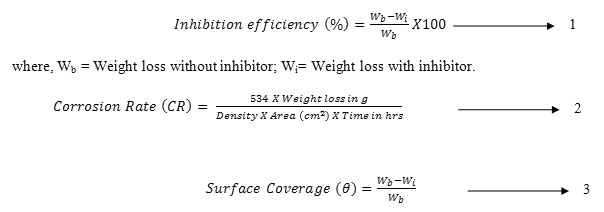
where, Wb = Weight loss without inhibitor; Wi= Weight loss with inhibitor.
Computational Details
B3LYP, a version of the DFT method that uses Becke’s three parameter functional (B3) and includes a mixture of HF with DFT exchange terms associated with the gradient corrected correlation functional of Lee, Yang and Parr (LYP) (11), was used to carry out quantum calculations. Full geometry optimization together with the vibrational analysis of the optimized structures of the inhibitor was carried out at the B3LYP/6-31G (d) level of theory using G03W program package. The quantum chemical parameters were calculated for molecules in non-protonated as well as in the protonated form in aqueous form. It is well known that the phenomenon of electrochemical corrosion occurs in liquid phase. As a result, it was necessary to include the effect of a solvent in the computational calculations. In the G03W program, SCRF methods (Self-consistent reaction field) were used to perform calculations in aqueous solution. These methods model the solvent as a continuum of uniform dielectric constant and the solute is placed in the cavity within it.
There is no doubt that the recent progress in DFT has provided a very useful tool for understanding the molecular properties and for describing the behaviour of atoms in molecules. DFT methods have become very popular in the last decade due to their accuracy and less computational time.Beside the geometries of the compounds, an analysis of quantum chemical parameters provided valuable information on the reactivity and selectivity of azole derivatives. These information are valuable in selecting a suitable compound or compounds (among compounds of similar structural features) to use as corrosion inhibitor as they inform which molecule has greater tendency to donate electrons, receive electrons or bind more strongly to the metal surface. Quantum chemical parameters such as the energy of the Highest Occupied Molecular Orbital (EHOMO) and the energy of the Lowest Unoccupied Molecular Orbital (ELUMO), the energy difference (ΔE) [EHOMO – ELUMO], the dipole moment (D), the charges on the atoms are often sighted among the most important quantities that provide information on the reactivity of the systems under consideration. Other quantities include the hardness (η), softness (S), electrophilicity index (ω) and the fractions of electrons transferred (ΔN). The various parameters are collectively reported in Table 3.
According to Koopman’s theorem(12, 13)the ionization potential (I) and electron affinity (A) of the inhibitors are calculated using the following equations.
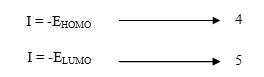
The higher HOMO energy corresponds to the more reactive molecule in the reactions with electrophiles, while lower LUMO energy is essential for molecular reactions with nucleophiles
Electronegativity (χ) is the measure of the power of an electron or group of atoms to attract electrons towards itself (14) and is estimated using the following equation

Global hardness (η) measures the resistance of an atom to a charge transfer [13] and is obtained from the equation

Global softness (σ) describes the capacity of an atom or group of atoms to receive electrons (13) and is the inverse of global hardness. It is estimated by using the equation

Electronegativity, hardness and softness have proved to be very useful quantities in the chemical reactivity theory.According to Pearson theory(15) the fraction of transferred electrons (ΔN) from the inhibitor molecule to the metallic atom can be calculated. For a reaction of two systems with different electronegativity (as a metallic surface and an inhibitor molecule), the following mechanism will take place: the electronic flow will occur from the molecule with the lower electronegativity toward that of higher value, until the chemical potentials are the same. For the calculation the following formula was used. Thus the fraction of electrons transferred from the inhibitor to metallic surface, ΔN, is given by
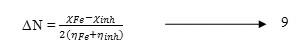
Where χFe and χinh denote the absolute electronegativity of iron and inhibitor molecule respectively ηFe and ηinh denote the absolute hardness of iron and the inhibitor molecule respectively. In order to calculate the fraction of electrons transferred, the theoretical value for the electronegativity of bulk iron was used χFe=7.0 eV(16)and a global hardness of ηFe = 0 by assuming that for a metallic bulk I= A(17). The difference in electronegativity drives the electron transfer, and the sum of the hardness parameters acts as a resistance.
Results and Discussion
Weight Loss Measurements
Corrosion inhibition efficiency of the inhibitors (azoles) calculated by weight loss measurements after 3hours of immersion time at 303 K are listed in Table 2. The data in Table 2 reveal that inhibition efficiency increases with an increase in concentration for each inhibitor (0.05 mM to 1 mM). The increase in inhibition efficiency with increasing concentrations of inhibitors is due to an increase in the surface coverage, resulting in retardation of metal dissolution (18). The anodic dissolution of iron in acidic and the corresponding cathodic reaction has been reported as follows (19).
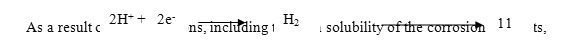
the metal loses weight in the solution. Experimentally, the inhibition efficiencies of the studied inhibitors followed the order PPP > POP.Maximum efficiency of 91.99% was observed for PPP. The corrosion rates were much less in the presence of inhibitors(20) as compared in the absence of inhibitors. The decrease in corrosion rate and high inhibition efficiency (%) of the inhibitors could be attributed to the adsorption of the entire inhibitor molecule onto the mild steel surface resulting in the formation of a protective adsorption film, which separates it from the corrosive medium. In the inhibited solutions, the corrosive rate is indicative of the number of free corroding sites remaining after some sites have been effectively blocked by the adsorption of the inhibitor (21). The effectiveness of the inhibitors for corrosion protection is mainly due to the presence of hetero atoms (O & N) and aromatic rings.
2Quantum Chemical Study of Non-Protonated form of the Studied Inhibitors in Aqueous Phase
The calculated quantum chemical descriptors provide trends in the reactivity and selectivity features of the studied compounds. Calculations were done in aqueous phase and by considering both the protonated and the non-protonated species. A comparison of the protonated and the non-protonated species show that there are some significant little differences in the quantum chemical parameters observed between the protonated and the non-protonated species.
Frontier orbital theory was useful in predicting the adsorption centers of the inhibitor molecules responsible for its interaction with the surface metal atoms (22). According to the frontier molecular orbital theory (FMO) of chemical reactivity, the formation of a transition state is due to the interaction between HOMO and LUMO levels of the reacting species(23). The smaller the orbital energy gap (ΔE) between the participating HOMO and LUMO levels, the stronger will be the interactions between the two reacting species. EHOMO is a quantum chemical parameter, which is often associated with the electron donating ability of the molecule. High values of EHOMO are likely to indicate a tendency of the molecule to donate electrons to appropriate acceptor molecules to the unoccupied d-orbital of a metal. As we know the electronic configuration of Fe atom is [Ar] 4s23d6 and 3d orbital is not fully filled with electrons. The unfilled 3d orbital could bind with HOMO of the inhibitors, whereas the filled 4s orbital could donate the electron to LUMO of the inhibitors. So, it can be predicted that the adsorption of inhibitors on the mild steel surface may be ascribed to the interaction between 3d,4s orbital of Fe atom and the front molecular orbitals of the inhibitor. Therefore, the energy of the lowest unoccupied molecular orbital (ELUMO) indicates the ability of the molecule to accept electrons(24). So, lower the value of ELUMO, the more probable the molecule would accept electrons. Thus the binding ability of the inhibitor to the metal surface increases with an increasing in the HOMO and decrease in the LUMO energy values. According to Table 3, the values of EHOMO follow the order, PPP> POP which correlate with the experimentally determined inhibition efficiency. The values of ELUMO follow the order, PPP < POP which correlates with the order of inhibition efficiency obtained experimentally.
The energy gap, ΔE = (ELUMO–EHOMO), is an important parameter and it is a function of reactivity of the inhibitor molecule towards the adsorption on metallic surface. As ΔE decreases, the reactivity of the molecule increases leading to increase in the inhibition efficiency of the molecule(25) The data from Table 3 indicate that the energy gap for the studied inhibitors follow the trend PPP< POP from which it can be concluded that PPP is the best inhibitor.
It is shown from the calculations that there was no obvious correlation between the values of the dipole moment with the trend of inhibition efficiency obtained experimentally. There is lack of agreement in the literature on the correlation between the dipole moment and inhibition efficiency(26, 27). It may be concluded that physical adsorption results from electrostatic interaction between the charged centers of the molecules and charged metal surface, which results in a dipole interaction of molecule and metal surface. Therefore, the positive sign of the coefficient µ suggests that these inhibitors can be adsorbed on the mild steel surface by physical mechanism (28).
Absolute hardness (η) and softness(S) are the important properties to measure the molecular stability and reactivity. A hard molecule has a large energy gap and a soft molecule has a small energy gap. Soft molecules are more reactive than hard ones because they could easily offer electrons to an acceptor. For the simplest transfer of electrons, adsorption could occur at the part of the molecule where σ, which is a local property, has the highest value(29). In a corrosion system, the inhibitor acts as a Lewis base while the metal acts as a Lewis acid. Bulk metals are soft acids and thus soft base inhibitors are most effective for acidic corrosion of these metals. It is shown from the calculations that PPP has the highest softness and the lowest hardness. Normally, the inhibitor with the least value of global hardness and highest value of global softness is expected to have the highest inhibition efficiency(30).
The number of electrons transferred (ΔN) was also calculated and presented in Table 3. Values of ΔN show that the inhibition efficiency resulting from electron donation agrees with Lukovits’s study(31). If ΔN < 3.6, the inhibition efficiency increases by increasing electron-donating ability of these inhibitors to donate electrons to the metal surface and it increases in the following order: PPP> POP. The results indicate that ΔN values correlate with experimental inhibition efficiencies trend. The optimized geometries, HOMO and LUMO of the two azole derivatives in non-protonated form in aqueous phase are shown in Table 6.
Quantum chemical study of protonated form of the studied inhibitors in aqueous phase
Organic inhibitors under investigation have a great tendency to be protonated in acidic medium due to the presence of N and O atoms. This is confirmed from the calculations which show the greater stability of protonated inhibitors (Table 4). It is shown from the optimized structures of the investigated inhibitors that there are one or more active centers on the inhibitor for protonation.
Most of the quantum chemical parameters/descriptors calculated in the protonated form such as total energy, EHOMO, ELUMO, energy gap (ΔE), hardness, softness and fraction of electrons transferred, were in accordance with the order of inhibition efficiency obtained experimentally PPP > POP.
Comparison of quantum chemical calculations for protonated and non-protonated inhibitors indicates that there is a clear correlation between most of the quantum chemical parameters/descriptor in the protonated form and non-protonated form of the studied azole derivatives in aqueous phase (Table 3 &4).
Table 4: Calculated quantum chemical parameters for the inhibitors in the protonated form obtained using DFT at the B3LYP/6-31G (d) basis set in aqueous phase.
|
Quantum chemical parameters |
PPP |
POP |
| Total energy (eV) | -706.68 | -726.52 |
| Dipole moment (debye) | 9.0532 | 11.3860 |
| EHOMO (eV) | -3.1217 | -3.5141 |
| ELUMO (eV) | -1.8368 | -1.6243 |
| ΔE gap (eV) | 1.2849 | 1.8898 |
| Ionization potential (eV) | 3.1217 | 3.5141 |
| Electron affinity (eV) | 1.8368 | 1.6243 |
| Electronegativity (eV) | 2.4793 | 2.5692 |
| Global hardness (eV) | 0.6425 | 0.9449 |
| Global softness | 1.557 | 1.0583 |
| ΔN | 3.5184 | 2.3446 |
Mulliken Charge Density Distribution
The more negative the atomic charges of the adsorbed center, more easily the atom can donates its electrons to the unoccupied orbital of the surface of the metal atoms and more easily the electrostatic attraction between the surface and the studied molecules. The Mulliken charge distributions for non-protonated and protonated forms are presented in Table 5. In both non-protonated and protonated forms, nitrogen and oxygen atoms have higher charge densities. The regions of the highest electron density are generally the sites to which electrophiles attacked. Therefore N and O atoms are the active centers, which have the strongest ability of bonding to the metal surface. From the molecular orbital density distribution, Tables 6 &7, it can be recognized that, the electron density of the frontier orbital is proportioned over several atoms. With this kind of structure, it is difficult to form chemical bond with active centers, which proves the probability of the physical adsorption between the interaction sites.
Table 5: Calculated Mulliken atomic charges for the inhibitors in non-protonated and protonated form obtained using DFT at the B3LYP/6-31G (d) basis set in aqueous phase.
|
Non-protonated |
Protonated |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
PPP |
POP |
PPP |
POP |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
Atoms |
Atoms |
Atoms |
Atoms |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|
|
|
|
|
N1 O2 C3 C4 C5 C8 C9 C10 C13 C14 C17 C20 C21 C22 N23 C24 C25 |
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
 |
Table 6: The 3D-structure of synthesized inhibitors in the neutral form obtained using DFT at the B3LYP/6-31G (d) basis set in aqueous phase. Click here to View table |
 |
Table 7: The 3D-structure of synthesized inhibitors in the protonation form obtained using DFT at the B3LYP/6-31G (d) basis set in aqueous phase. Click here to View table |
Mechanism of Inhibition
The corrosion inhibition of the azole derivatives is mainly attributed to the adsorption on mild steel surface. These compounds can be adsorbed in a flat orientation through tridentate (azole) form. The surface coordination is mainly through the nitrogen and oxygen active centers. The mode of adsorption depends on the affinity of the metal towards π-electron cloud of the ring system. Metals such as Cu and Fe that have a greater affinity towards aromatic moieties were found to adsorb benzene rings in a flat orientation.
The adsorption of these inhibitors on mild steel surface may take place in the following ways(29): (i) the inhibitor molecules may be adsorbed via donor-acceptor interactions between the πelectrons of the aromatic rings and unshared electron pairs of the heteroatoms to form a bond with the vacant d-orbitals of the metal surface (chemisorption). (ii) In acidic media, the N heteroatoms are readily protonated, which might adsorb onto the metallic surface via the negatively charged acid anion ( ) (physisorption). Thus, physical and chemical adsorption will lead to the formation of protective films of the inhibitor molecules onto the steel surface.
Conclusions
- The investigated heterocyclic azole derivatives show good inhibition efficiencies for the corrosion of mild steel in H2SO4 solution.
- The inhibition efficiency increases with an increase in concentrations of the inhibitor. The order of inhibition as follows: PPP > POP.
- Computed quantum chemical properties such as EHOMO, ELUMO, energy gap (ΔE), dipole moment (µ), hardness (η), softness (S), the fractions of electrons transferred (ΔN) were found in good correlation with experimentally determined inhibition efficiency in both non-protonated and protonated form.
References
- Saranya, J., Sounthari, P., Kiruthika, A., Saranya, G., Yuvarani, S., Parameswari, K., Chitra, S, Orient. J. Chem.2014, 30, 1719-1736.
- Saranya, J., Sounthari, P., Kiruthuka, A., Parameswari, K., and Chitra, S, Orient. J. Chem., 2014, 30, 1971-1987.
- Ju, H.; Kai, Z. P.; Li, Y. Corros. Sci.2008, 50, 865-871
- Liu, B.; Xi, H.; Li, Z.; Xia,Q.Appl. Surf. Sci.2012, 258, 6679-6687
- Gece, G.Corros.Sci. 2008, 50, 2981-2992
- Bereket, G.; Ogretir,C.; Yurt,A.J. Mol. Struct. (THEOCHEM),2001, 571, 139-145
- Awad, M. K.J. Electroanal. Chem.2004, 567, 219-225
- Awad,M.K.; Mahgoub,F.M.; El-iskandrani,M.M.J. Mol. Struct. (THEOCHEM), 2000, 531, 105-117
- Obot, I.B.; Obi-Egbedi, N.O.Corros. Sci.2010, 52, 657-660
- Abbas, A.F.; Turki, A.A.; Hameed, A.J.J. Mater. Environ. Sci.2012,3, 1071-1078
- Lee, C.; Yang, W.; Parr, R.J.Phys. Rev.1988, B37, 785-789
- Geerlings, P.; Proft, F. De.; Langenaeker, W. Chem. Rev.2003, 103, 1793-1874
- Parr, R.G.; Pearson,R.G.;J. Am. Chem. Soc.1983, 105, 7512-7516.
- Pauling, L., The nature of the chemical bond (Cornell University Press, Ithaca, New York, (1960)
- Pearson, R.G.; Inorg. Chem.1988, 27, 734-740
- Sastri, V. S.; Perumareddi, J. R.Corros. Sci.1997, 53, 617-621
- Dewar,M. J. S.; Thiel,W.J. Am. Chem. Soc.1977, 99, 4899-4907
- Ezeoke,A.U.; Adeyeml,O.G.; Akerele,O.A.; Obi Egbedi,N.O.Int. J. Electrochem. Sci.2012, 7, 534-553
- Amin M.A.; Khaled, K.F. Corros. Sci.2010, 52, 1762-1770
- Kumar, S.; Sharma, D.; Yadav, P.; Yadav, M.Ind. Eng. Chem. Res.2013, 52, 14019-14029
- Arukalam,I. O.; Madu, I. O.; Ijomah,N. T.; Ewulonu,C. M.; Onyeagoro,G. N.Journal of materials,2014, 1-11
- Kandemirli, F.; Sagdinc, S.Corros. Sci.2007, 49, 2118-2130
- Musa, A.Y.;Kadhum, A.A.H.;Mohamad, A.B.; Rahoma, A.A.B.;Mesmari, H.J. Mol. Struct.2010, 969, 233-237
- Ahamad, I.; Prasad, R.; Quraishi,M.A.Corros. Sci.2010, 52, 1472-1481
- Saranya, J., Sounthari, P., Kiruthuka, A., Parameswari, K., and Chitra, S, J. Mater. Environ. Sci., 2015, 6, 425-444.
- Stoyanova, A.; Petkova, G.;Peyerimhoff, S.D.Chem. Phys.2002, 279, 1-6
- Rodriguez-Valdez,L.M.; Martinez-Villfane, A.; Glossman-Mitnik, D. J. Mol. Struct.(THEOCHEM), 2005,713, 65-70
- Khalil, K.F. Electrochim. Acta. 2003,48, 2635-2640
- Hasanov, R.; Sadikoglu, M.; Bilgic, S.Appl. Surf. Sci.2007, 253, 3913-3921
- Ebenso, E.E.;Isabirye, D.A.; Eddy,N.O. Int. J. Mol. Sci.2010, 11, 2473-2498
- Lukovits, I.; Kalman, E.;Zucchi, F.Corrosion(NACE), 2001, 57, 3-8

This work is licensed under a Creative Commons Attribution 4.0 International License.
![]()
A New Edition of Web of Science
Journal Impact Factor
2022: 0.5
Five Year: 0.8
Journal is Indexed in

Cabells Whitelist
![]()


